Transition Metals
2p – Multiplet Splitting
due to Unpaired Electrons in Valence Levels
Cr (3+), Mn (3+), Fe (3+), Co (3+), Ni (2+), Cu (2+)
MULTIPLET SPLITTING (split final state) is due to Interaction of Unpaired Electrons in Valence Levels and Unpaired Core Electron
by Mark C. Biesinger
Multiplet Splitting occurs in core level XPS whenever there is one (or more) unpaired electron(s) in the valence levels. Multiplet splitting occurs due to the exchange interaction between the unpaired valence electrons and the unpaired electron left in the core level (after photoionization). This interaction produces “split final states”.
In other words:
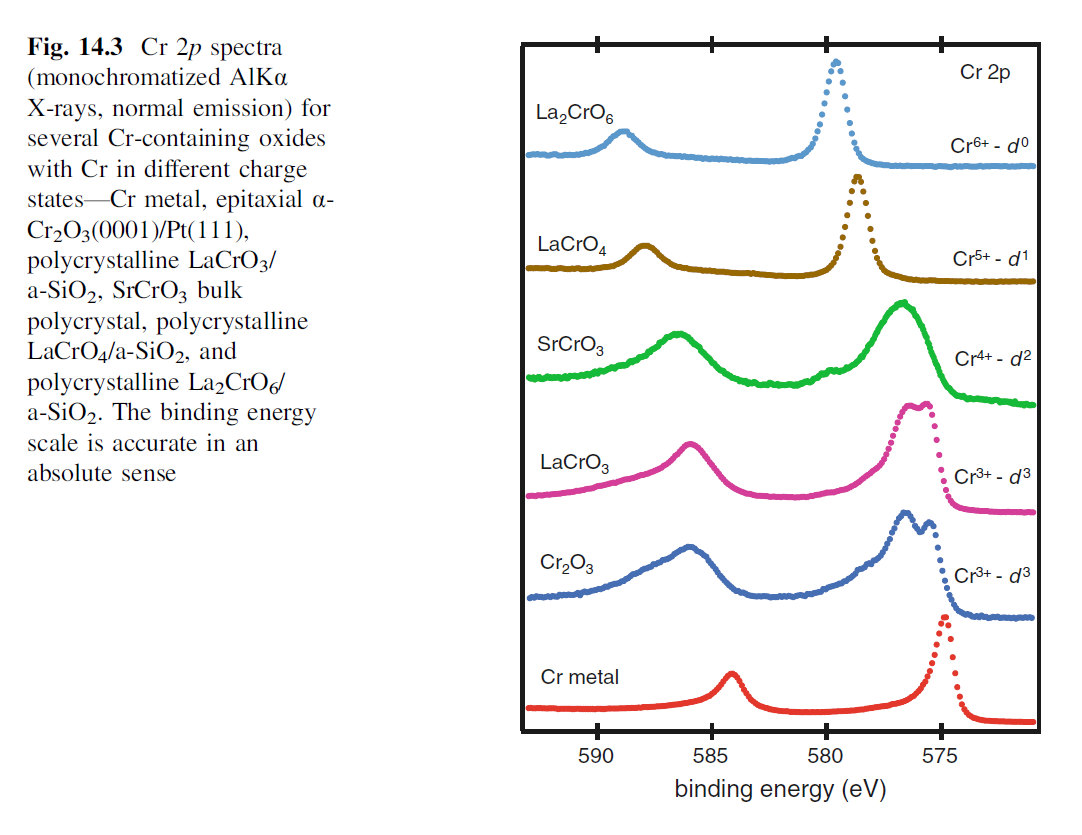
Multiplet splitting arises when an atom contains unpaired valence electrons. When a core electron vacancy is created by photoionization, there can be coupling between the remaining unpaired electron in the core with the unpaired electrons in the outer shell. This can create a number of final states, which will be seen in the photoelectron spectrum as a multi-peak envelop. The Figure below (Cr 2p spectrum of Cr2O3) shows the multiplet splitting structure that exists for the Cr 2p3/2 peak for a fractured Cr2O3 single crystal.


Multiplet Splitting (split final states) occurs for compounds having unpaired valence electrons interacting with unpaired electron in core – after photoionization:
- 1s, 2s, or 3s electrons in materials such as: O2 gas, CO gas, NO gas, NNO gas, MnO solid, MnF2 solid
- 2p, 3p, or 4p, electrons in materials such as CuO, CuSO4, etc
- 3d, or 4d electrons in rare earth elements
Discussion on Multiplet Splitting from paper by Paul Bagus
P.S. Bagus, E. Ilton and C. Nelin, Surface Science Reports, Vol 68, p273-304, 2013
In the present section we expand this earlier treatment to consider a wide variety of many-body effects that can lead to satellites, often with very large intensities. A suitable definition for many-body effects is that they require the use of wavefunctions that cannot be represented by a single configuration or a single CSF. In particular, for closed shell systems, this means that we must go beyond a single determinant description of the wavefunction. Final states that correspond to multiplets are also considered a many-body effect, although the reasoning requires explanation. A multiplet can be regarded as a degenerate set of states that arise from the angular momentum coupling of the open shells in a single configuration [88] and, hence, not a many-body effect. On the other hand, wavefunctions that are eigenfunctions of the orbital and spin angular momentum operators normally are combinations of determinants [88] and, hence, multiplets can be grouped with many body effects. Furthermore, there is a type of angular momentum coupling that is a pure many-body effect.
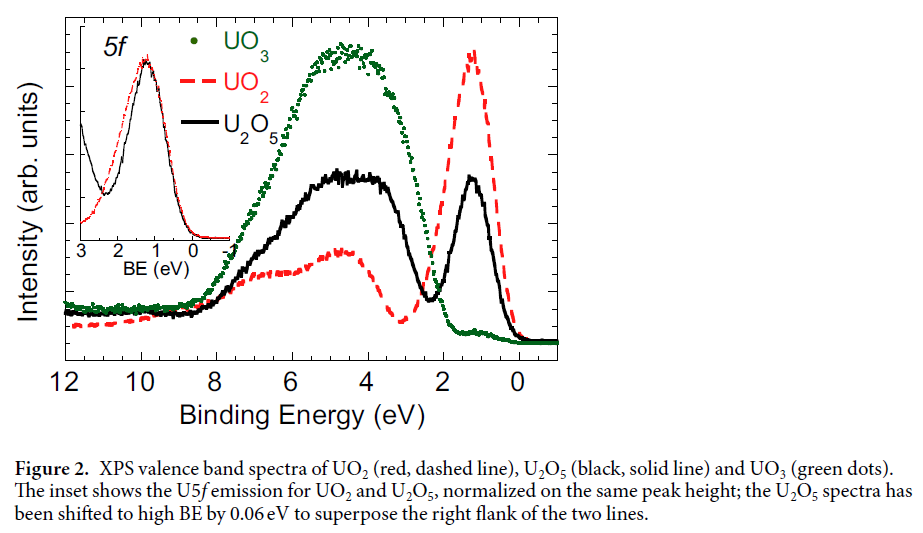
This occurs when the valence open shell can couple to multiplets other than the ground state multiplet of the initial state configuration. It is often possible to couple the core-hole with the re-coupled valence open shell to give a total multiplet the same as when the core-hole is coupled to the ground state multiplet of the valence open shell [30,166,167].While these angular momentum re-coupled multiplets are XPS forbidden, they are able to mix with the XPS allowed multiplets and thus lead to “satellites” with large intensity [30]. An example of the many-body mixing of XPS forbidden with XPS allowed multiplets in given in Section 6.2. For all these reasons, we chose to group the discussion of XPS multiplets with many body effects. In Section 6.1, we consider the multiplet splitting of the N(1s) and O(1s) XPS of NO [17]. We also examine the Mn 3s XPS [4], where multiplet theory is not sufficient. In Section 6.2, angular momentum recoupling is introduced as a major correction to the simple angular momentum coupling theory. This recoupling is important because the re-coupled CSFs are nearly degenerate with the XPS allowed multiplets. Other types of near degeneracy are also discussed that provide a more complete description of “atomic” processes than multiplet theory alone. In this section, the effects largely depend on orbitals with dominantly atomic character but which can be modified by ligand field splittings and covalent bonding. In Section 6.3, we consider very recent results [21,22,52,53] for the XPS of the two closed shell oxides, CeO2 and UO3 whose covalent character was discussed in Section 5.
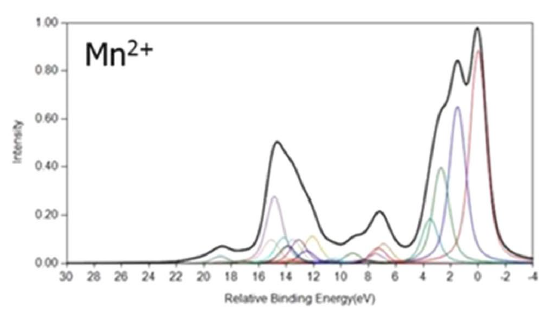
This small deviation from the statistical is relevant for the Mn 3s XPS multiplet splitting, discussed next. Other examples of multiplet splitting in molecules can be found in Ref. [6]. The dominantly ionic character of MnO, Section 5, where the 5 open shell electrons are ∼90% Mn 3d, even for a deep 2p core-hole, suggests that an isolated Mn2+ cation with a 3d5 open shell coupled to the high spin, 6S, multiplet would be a suitable model for the Mn XPS. A direct comparison of the measured 3s XPS in Mn atoms and in MnF2 and MnO crystals shows that the spectra are very similar for all these systems, which justifies the belief that Mn2+ is a good model for the XPS of MnO [169]. It should be noted that the atomic XPS data in Ref. [169] was for Mn atoms and not the Mn2+ cations appropriate for ionic crystals and that the energy separation of the first two peaks is slightly larger for Mn0 than for MnF2 and MnO. This slightly larger separation is reproduced in the calculated separation of the first two peaks between Mn0 [47] and Mn2+ [3].
One expects that the energy separations and the relative intensities of the 3s XPS peaks should be similar for Mn2+ and Mn0 since the 4s electrons, present in Mn0, are spectators for the core-level ionization, However, there will be some differences, which arise because a portion of the 4s charge density penetrates toward the core of the Mn atom. It is important that the differences between the theoretical results for Mn0 and for Mn2+ are mirrored in the differences between the XPS for the MnO or MnF2 crystals and the Mn atom. Following the logic for NO, discussed above, the 3s XPS should have two peaks. 7S and 5S split by 6K(3s,3d) [88]. HF theory predicts a splitting of ∼14 eV and an intensity ratio of 1.4:1 while the XPS measurements for several ionic crystals give a splitting of ∼6 eV with an intensity ratio of ∼2:1 [4,169]. This ratio is larger, by over 40%, than the statistical ratio of the two multiplets and is outside the range of deviation suggested from the results for NO; see above. The ∼8 eV error in the multiplet splitting is also much greater than would be expected from the errors of BEs calculated from atomic HF calculations [51,70]. Clearly, many-body effects other than multiplets must make important contributions as discussed in the following sub-section.
Table 1. First row transition metal species show multiplet splitting in their XPS spectra.
Biesinger Summary Table
![]()

Crist Summary Table
Peak-fitted spectra from Mark Biesinger at U. Western Ontario, Surface Science Western
Calculated Peak-shapes from Gupta and Sen, Physical Review, Vol 12, p15-18, 1975
2p Multiplet Splitting – Cr (3+), Mn (3+), Fe (3+), Co (3+), Ni (2+), Cu (2+)
| Peak-fits – Experimental Spectra Multiplet Splitting? – Present or Absent |
Peak-shapes – Calculated Multiplet Splitting by Gupta and Sen, Physical Review, vol 12, p15-18, 1975 |
|
| Chromium (Cr) Cr (2p) spectrum |
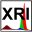
Multiplet Splitting – Experimental Cr (2p) spectrum – Cr2O3 Cr (3+) oxidation state [Ar] 3d54s1, 6 unpaired electrons Hi or Lo spin ? paramagnetic |
Multiplet Splitting – Calculated Cr (2p) spectrum – Gupta & Sen Cr (3+) oxidation state  |
| Chromium (Cr) |
NiCr2O4 [NiO-Cr2O3] (paramagnetic)
|
|
| Chromium (Cr) |
CrCl3 (anhydrous) (paramagnetic)
|
|
| Chromium (Cr) |
CrBr3 – 6H2O (diamagnetic)
|
|
| Chromium (Cr)
|
CrF3 (paramagnetic) Cr (3+) oxidation state [Ar] 3d54s1, ? unpaired s electron ? Hi or Lo spin ? 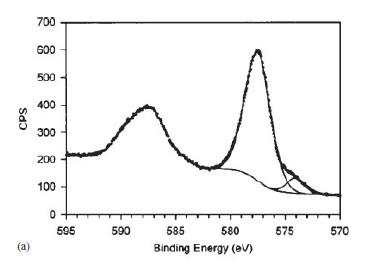 |
|
| MnO2 (paramagnetic) Mn (4+) oxidation state [Ar] 3d54s2, ? unpaired s electron ? Hi or Lo spin ?  |
 |
|
| Mn2O3 (paramagnetic) Mn (3+) oxidation state [Ar] 3d54s2, ? unpaired s electron ? Hi or Lo spin ?MnO (paramagnetic) Mn (2+) oxidation state [Ar] 3d54s2, ? unpaired s electron ? Hi or Lo spin ?  |
|
|
| Iron (Fe) | Fe2O3 (paramagnetic) Fe (3+) oxidation state [Ar] 3d64s2, 4 unpaired d electrons Hi or Lo spin ? 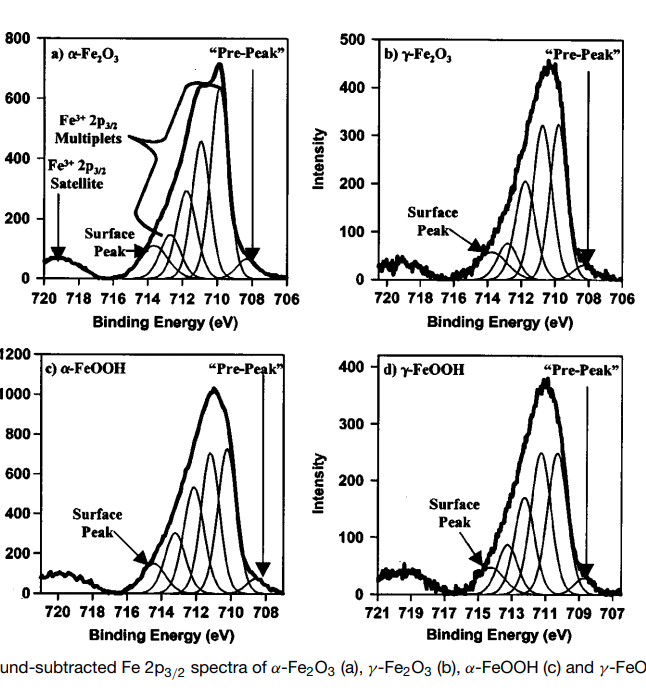 |
 |
| Fe3O4 – FeO-Fe2O3 (paramagnetic) Fe (3+) and Fe (2+) oxidation states [Ar] 3d64s2, 4 unpaired d electrons ? Hi or Lo spin ?  |
|
|
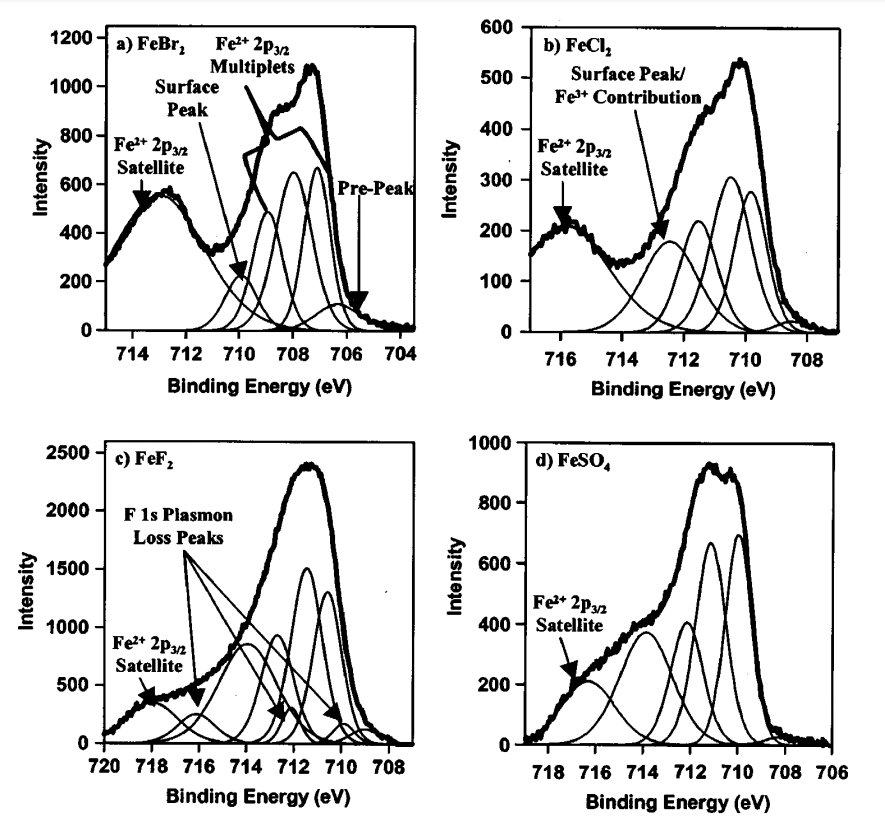 |
|
|
| FeBr3 FeCl3 FeF3 (paramagnetic) Fe (3+) oxidation state [Ar] 3d64s2, 4 unpaired d electrons ? Hi or Lo spin ?  |
|
|
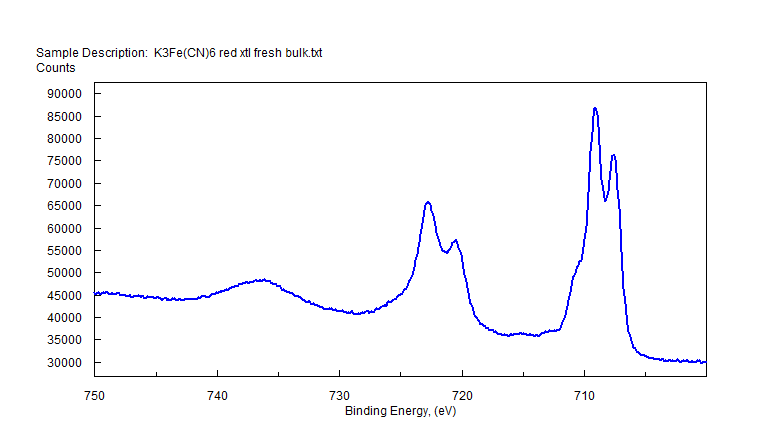
| Iron (Fe) | K4Fe(CN)6 (diamagnetic) Fe (4+) oxidation state ? [Ar] 3d64s2, 4 unpaired d electrons ? Hi or Lo spin ? 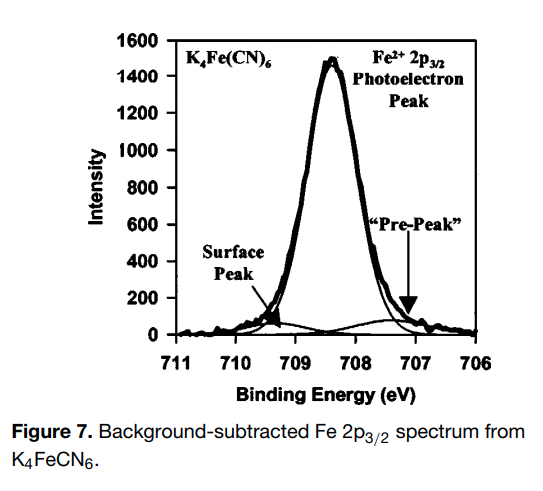 |
|
| Iron (Fe) | Fe (3+) vs Fe (2+)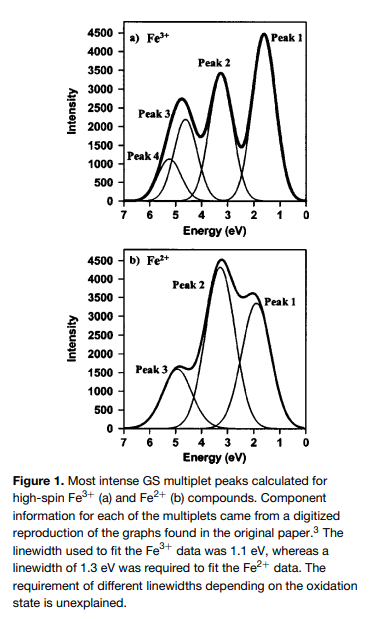 |
|
| Cobalt (Co) | Co3O4 – CoO-Co2O3 (paramagnetic) Co (3+) and Co (2+) oxidation states [Ar] 3d74s2, 3 unpaired d electrons ? Hi or Lo spin ?Co(OH)2 (paramagnetic) Co (2+) oxidation state [Ar] 3d74s2, 3 unpaired d electrons ? Hi or Lo spin ?CoO (paramagnetic) Co (2+) oxidation state [Ar] 3d74s2, 3 unpaired d electrons ? Hi or Lo spin ?  |
 |
| Nickel (Ni) | NiFe2O4 – NiO-Fe2O3 (paramagnetic) Ni (2+) oxidation state [Ar] 3d84s2, 2 unpaired d electrons ? Hi or Lo spin ?  |
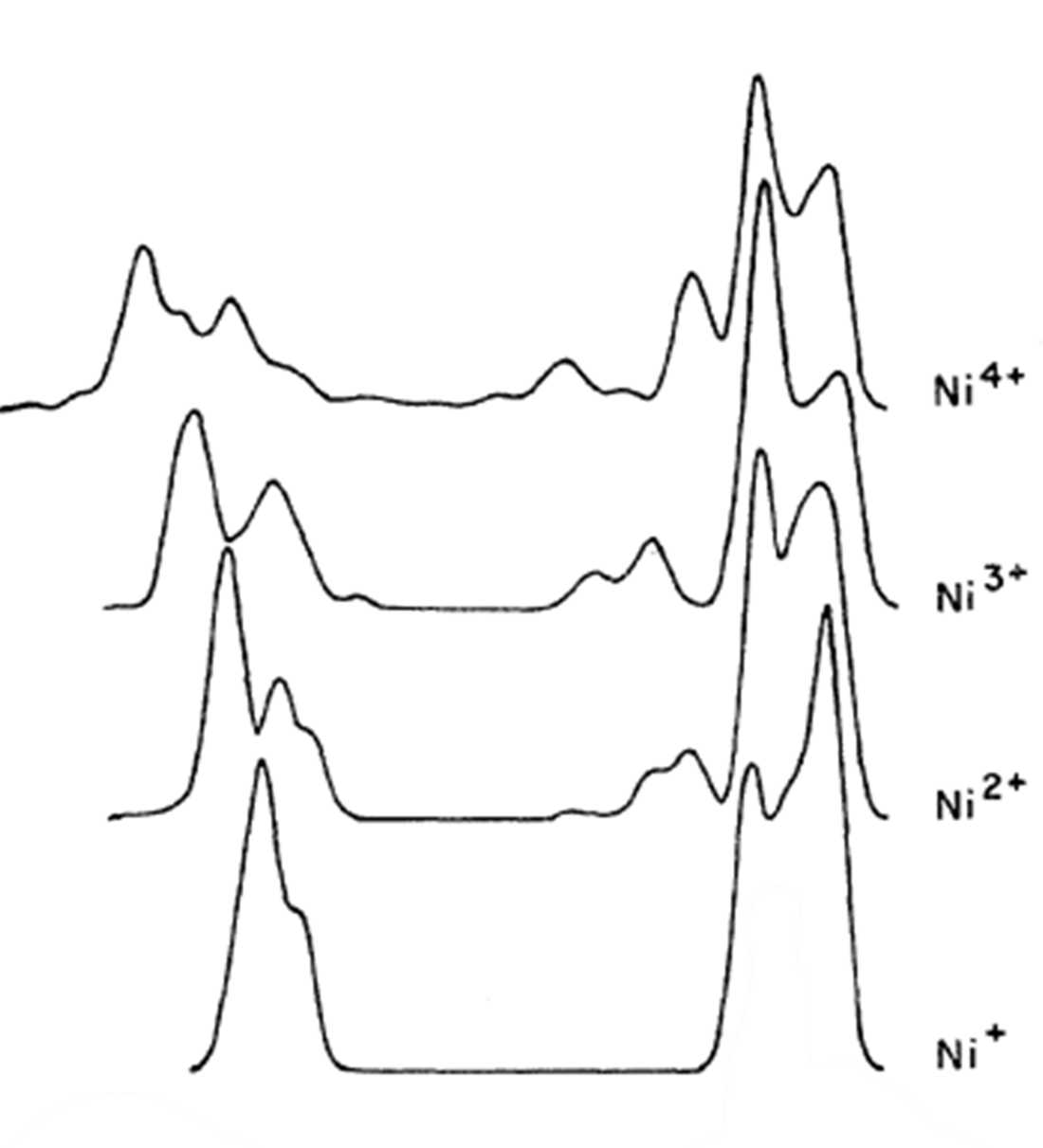 |



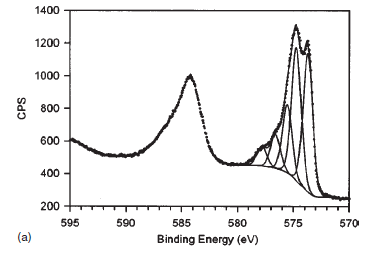

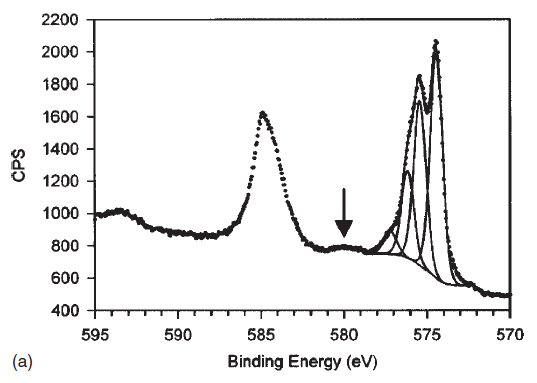

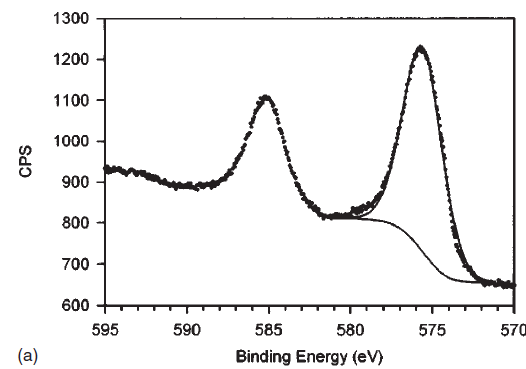




















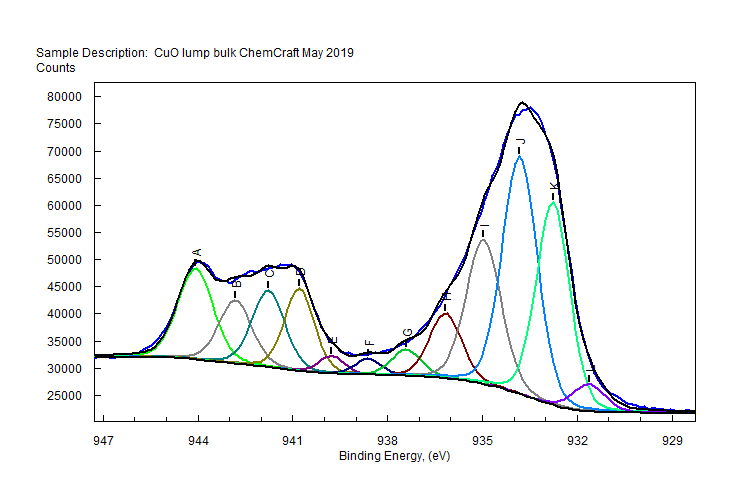
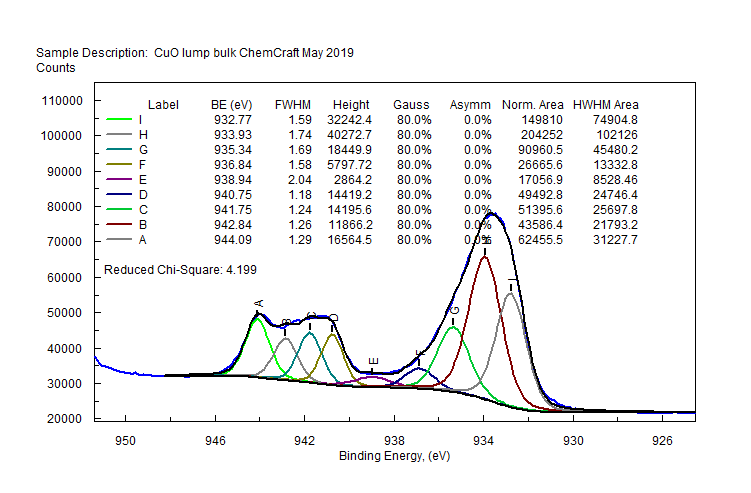
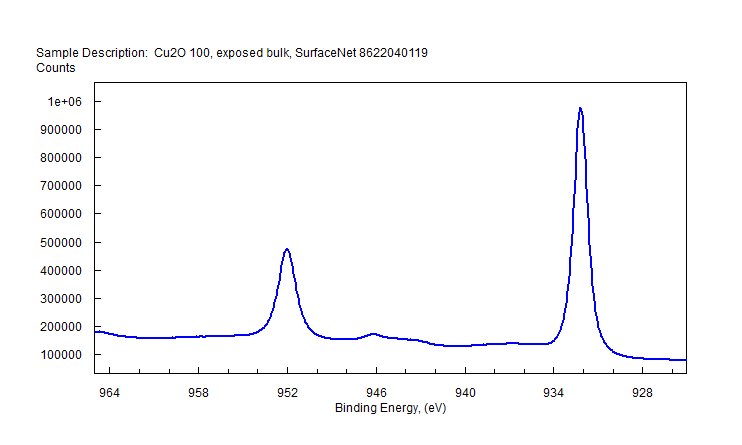
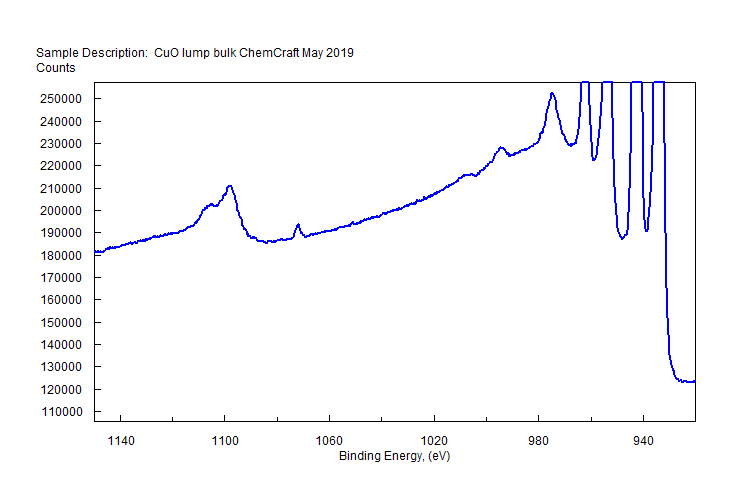
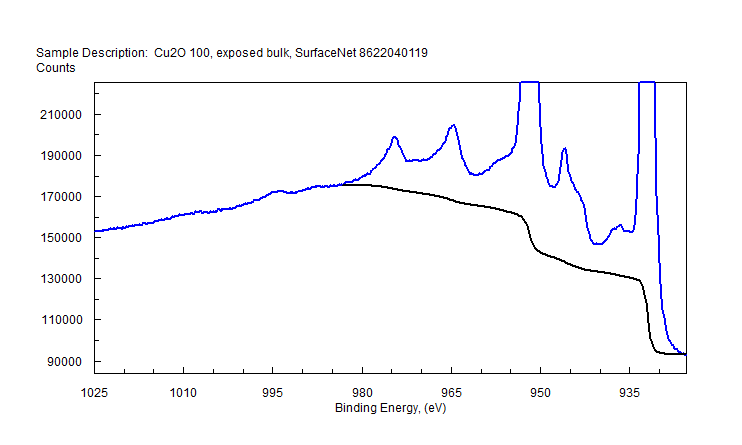
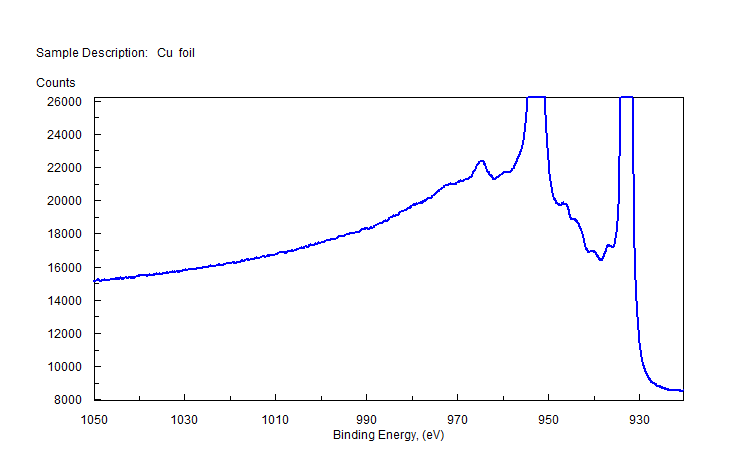
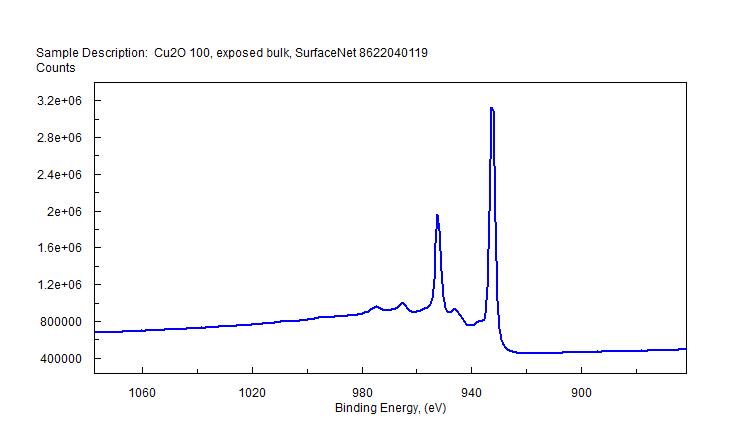
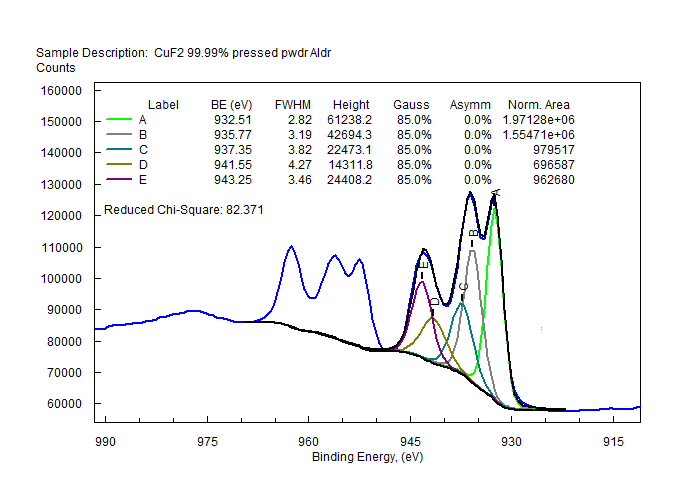
| Copper (Cu) | CuO (paramagnetic) Cu (2+) oxidation state [Ar] 3d104s1, 1 unpaired s electron Hi or Lo spin ?  |
 |


| Crist Series | ||
 |
|
|


 |
|
|
 |
|
|
 |
||


Iron (Fe) |
||
 |
|
|
|
|


 |
|
|
 |
|
|
 |
|
|



| Copper (Cu) by Crist | ||
 |
 |
|
 |
 |
|
  |
 |


 |
||

 |

Multiplet Splitting – 3s – Orbitals
MULTIPLET SPLITTING of “3s” Orbitals (split final states)
Multiplet Splitting occurs in core level XPS whenever there is one (or more) unpaired electron(s) in the valence levels. Multiplet splitting occurs due to the exchange interaction between the unpaired valence electrons and the unpaired electron left in the core level (after photoionization). This interaction produces “split final states”.
In other words:
Multiplet splitting arises when an atom contains unpaired valence electrons. When a core electron vacancy is created by photoionization, there can be coupling between the remaining unpaired electron in the core with the unpaired electrons in the outer shell. This can create a number of final states, which will be seen in the photoelectron spectrum as a multi-peak envelop. The Figure below shows the multiplet splitting structure that exists for the
Multiplet Splitting (split final states) occurs for compounds having unpaired valence electrons interacting with:
- 3s electrons in materials such as: MnO, MnF2, CrF2, …
- 2p electrons in materials such as: CuO, CuSO4…
- 3d electrons in rare earth compounds such as: CeO2…
- Gaseous compounds exhibit multiplet splitting of the 1s orbital, such as: NO, NNO…
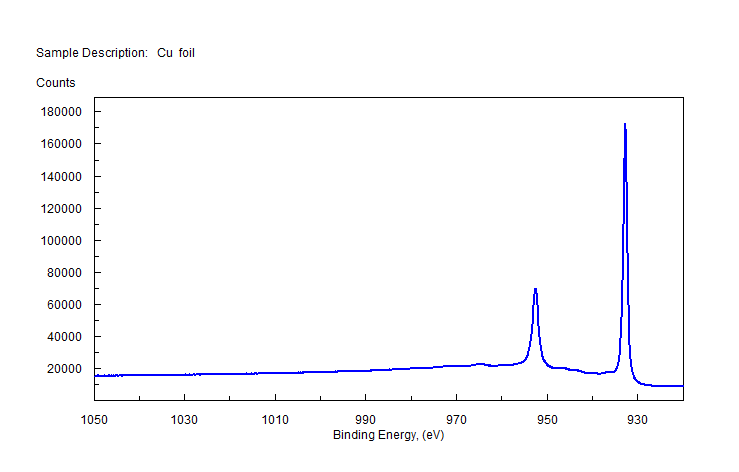
Pure Transition Metals – No “3s” Multiplet Splitting for Pure Metals
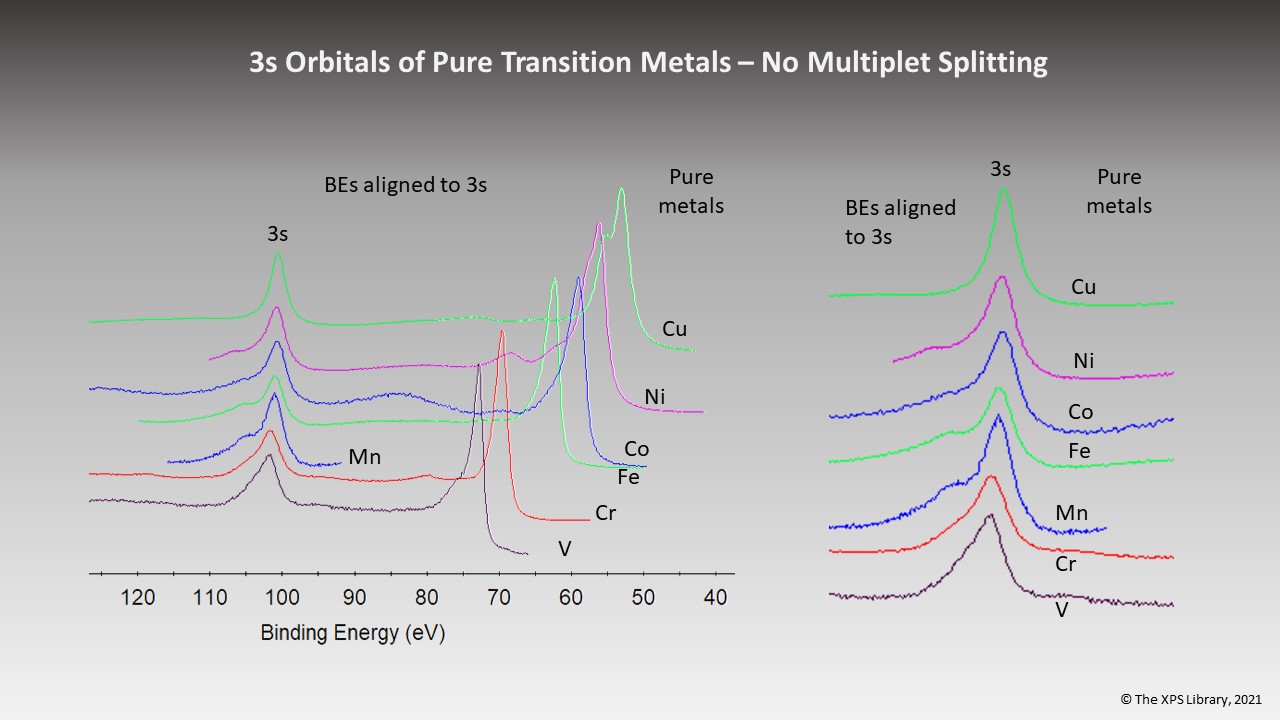

Electron Diagram for Multiplet Splitting of “3s” Orbitals in Chemical Compounds

Important Notes about “3s” XPS from Paul Bagus:
1. The TM 3s XPS is dominated by atomic effects. There are quantitative changes for metals and compounds but the basic physics is atomic physics.
2. The 3s XPS cannot be explained with a one-configuration model of multiplets. The Mn splitting from a simple exchange splitting model is 3X larger than observed; For other methods see the Viinikka paper.
3. The concept of FAC (Frustrated Auger Configuration) provides a powerful way to understand the many body effects that complicate the 3s XPS
4. Compelling proof of the character of the 3s XPS in magnetically ordered Fe was obtained from spin-polarized XPS; see paper with Mallow. It would be nice if the experiments could be extended to the region where the satellites are observed.
5. It is not possible to understand the 3s (or the 3p) XPS unless a many electron, many configuration model is used.
6. I view the spectra as arising from XPS forbidden configurations stealing intensity from the XPS allowed configurations. Pure theorists do not like this description because it isn’t perfect but it conveys the physics clearly and correctly.
7. The 3p XPS is even more complicated than the 3s XPS.
Spectra from Transition Metal Compounds – Having Multiplet Splitting in “3s” Orbital
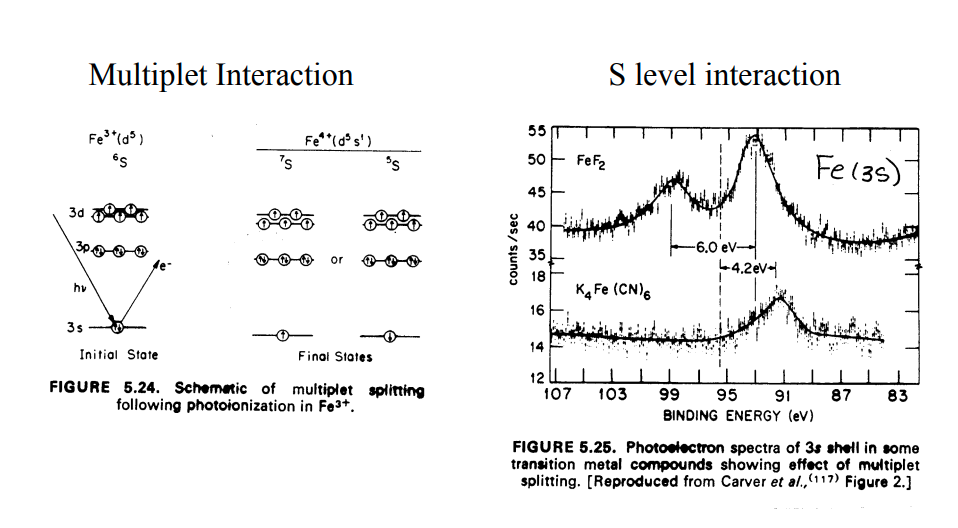

| Iron (3s) Final State Multiplet Splitting FeF2 solid Iron (Fe) atom electron configuration 1s2 2s2 2p6 3s2 3p6 3d6 4s2 2 un-paired electrons in 3d orbital |
||
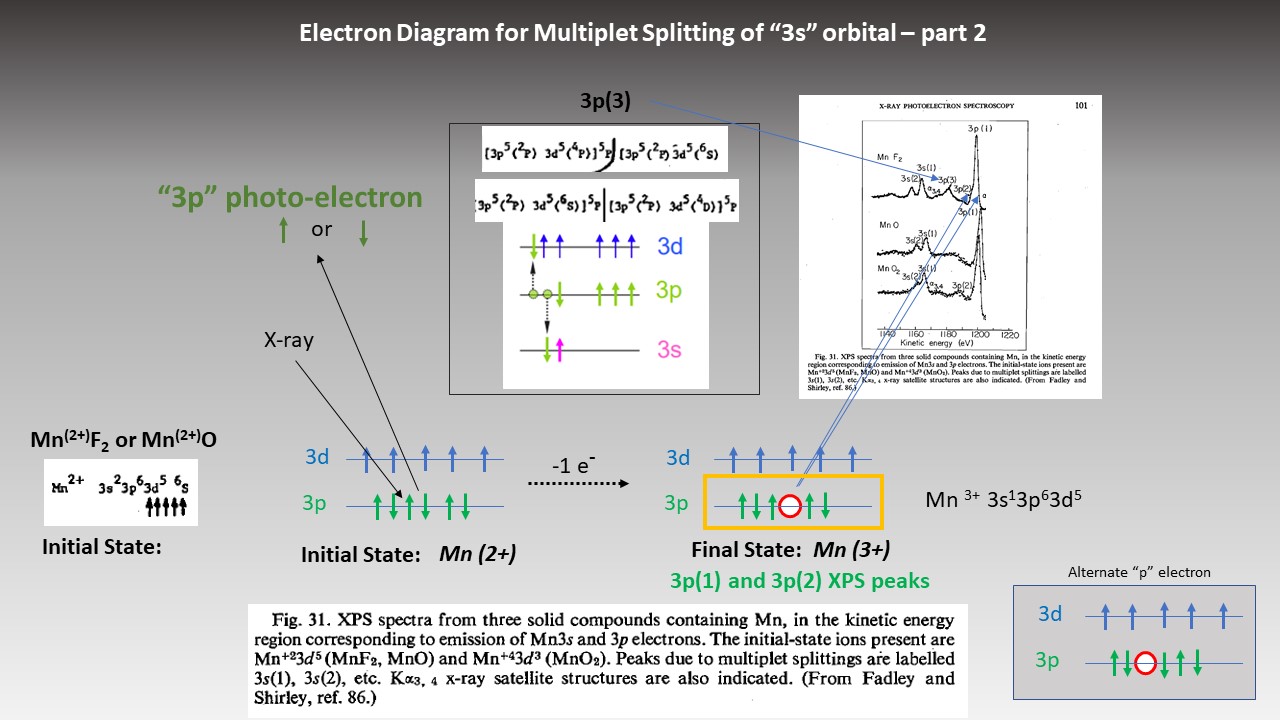
| Manganese (3s) Final State Multiplet Splitting MnO, MnF2, Mn (gas) Manganese (Mn) atom electron configuration 1s2 2s2 2p6 3s2 3p6 3d5 4s21 un-paired electrons in 3d orbital |
||
| Manganese (3s) Final State Multiplet Splitting MnF2, MnO, and MnO2 Manganese (Mn) atom electron configuration 1s2 2s2 2p6 3s2 3p6 3d5 4s21 un-paired electrons in 3d orbital |
||
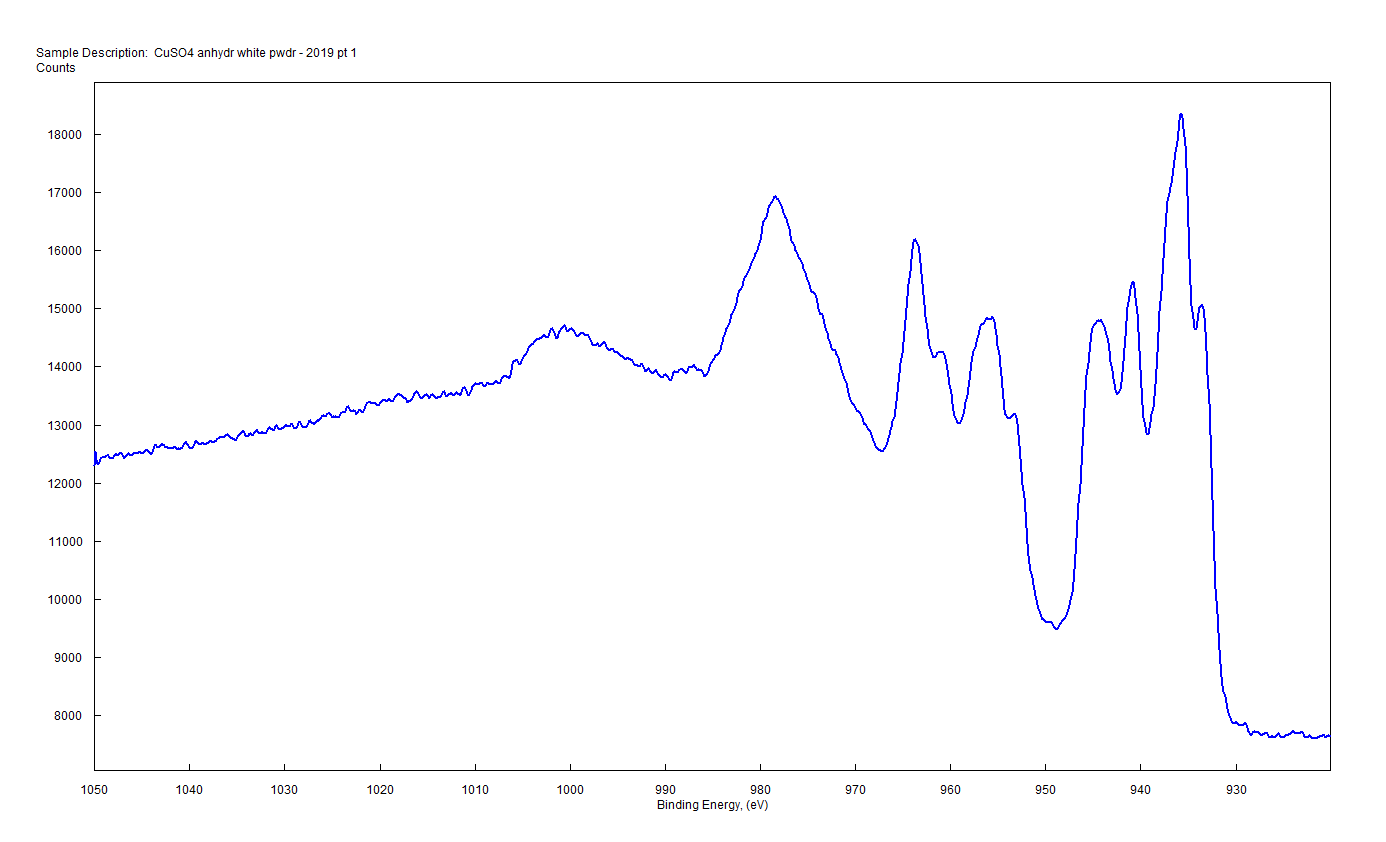
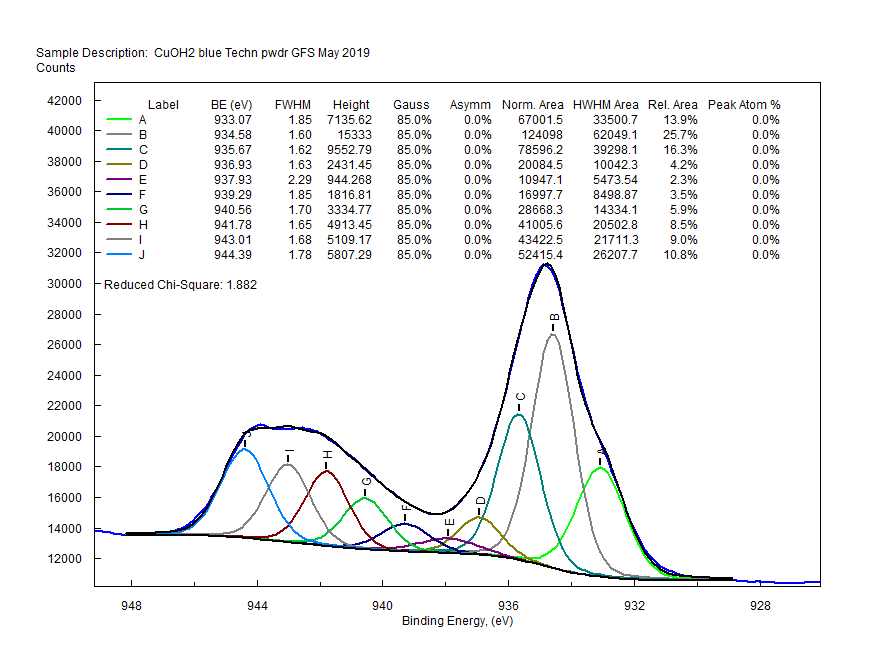
| Copper (3s) Final State Multiplet Splitting CuO solid Copper (Cu) atom electron configuration 1s2 2s2 2p6 3s2 3p6 3d10 4s11 un-paired electrons in 4s orbital |
|
|
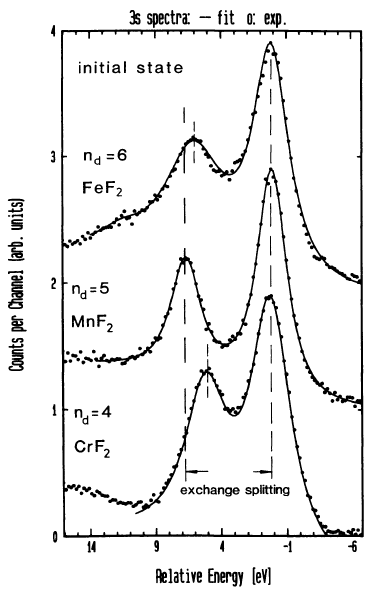
| Comparison of “3s” Multiplet Splittings
for FeF2, MnF2, and CrF2 |
 |
|




| Oxygen (1s) Final State Multiplet Splitting
Mixture of O2 gas and H2O gas Oxygen atom electron configuration 2 unpaired electrons O2 is Paramagnetic |
|
|
| Nitrogen (1s) Final State Multiplet Splitting
Nitrous Oxide, NO, gas Nitrogen atom electron configuration 1 unpaired electron NO is Paramagnetic |
 |



Rare Earths Compounds
Multiplet Splitting – La, Ce, Pr, Nd, Sm, Eu, Gd, Tb
Multiplet Splitting – La, Ce, Pr, Nd, Sm, Eu, Gd, Tb
 |
||
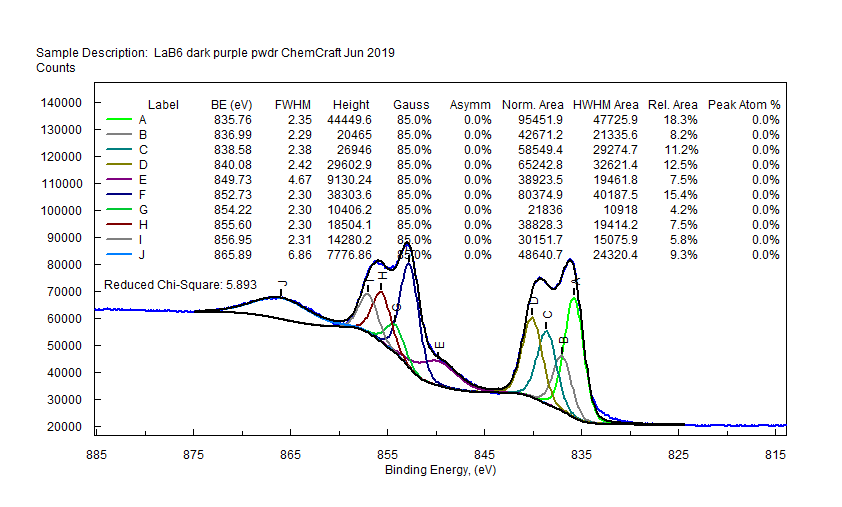 |
||
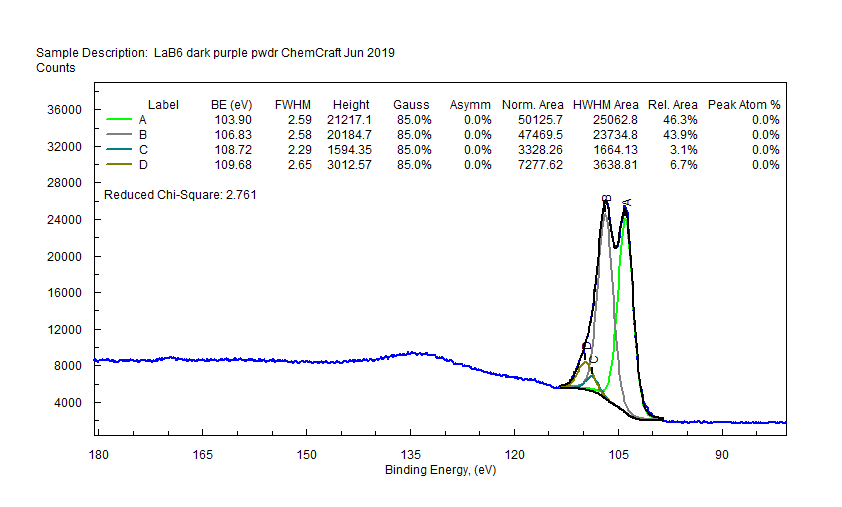 |
||
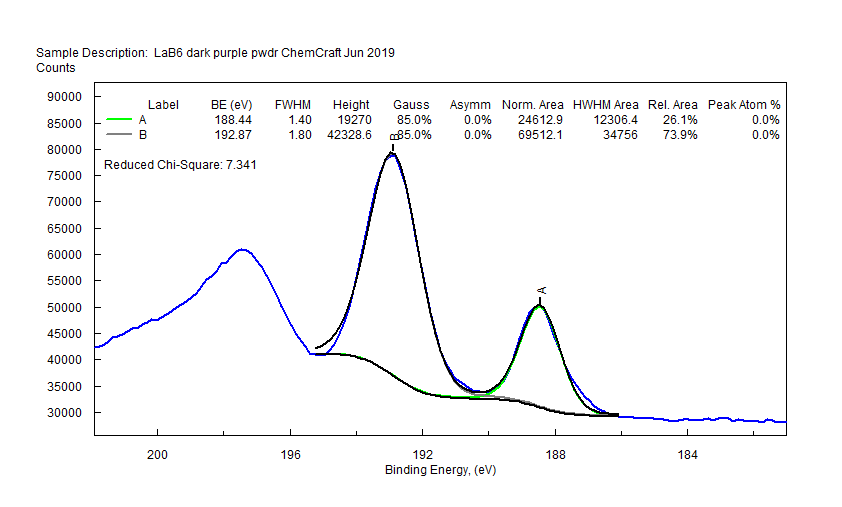 |
||
 |
||
 |
||
 |
||
 |
||
 |
||
 |


 |
||
 |
||
 |
||
 |
||
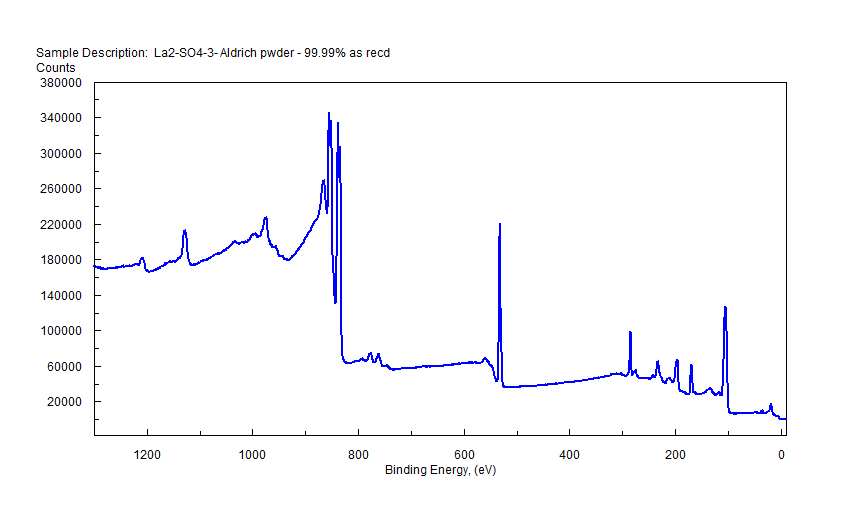 |
||
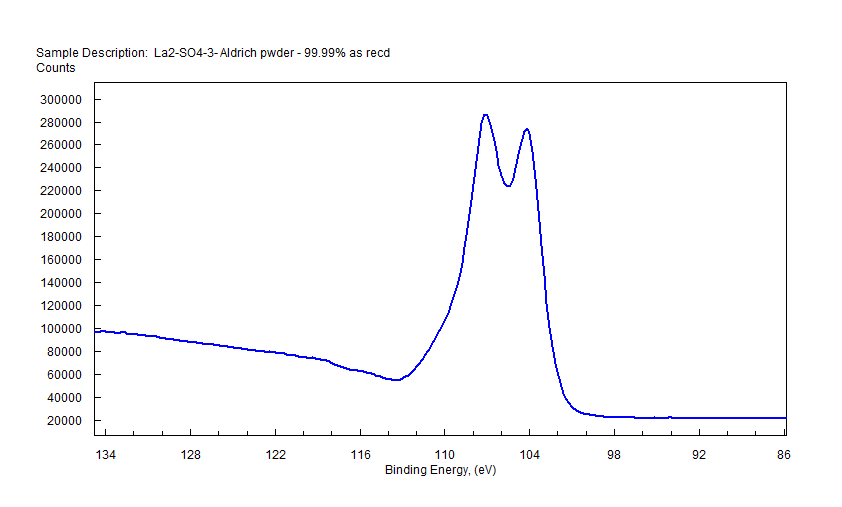 |
||
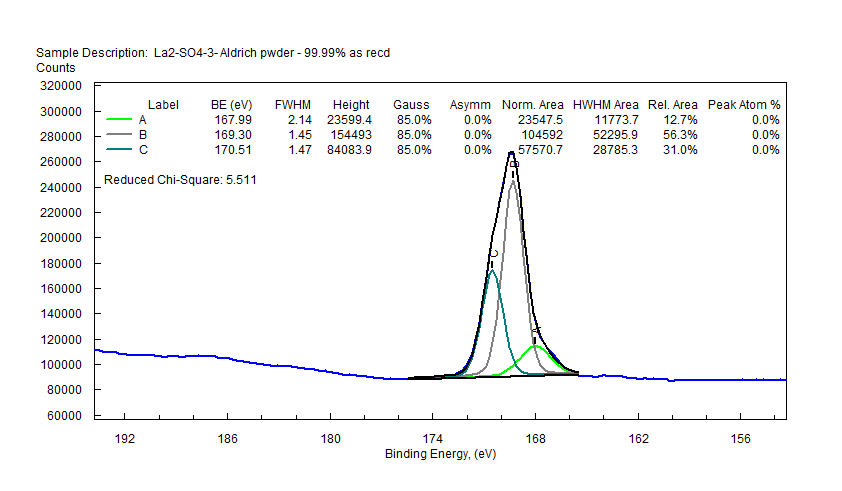 |
||
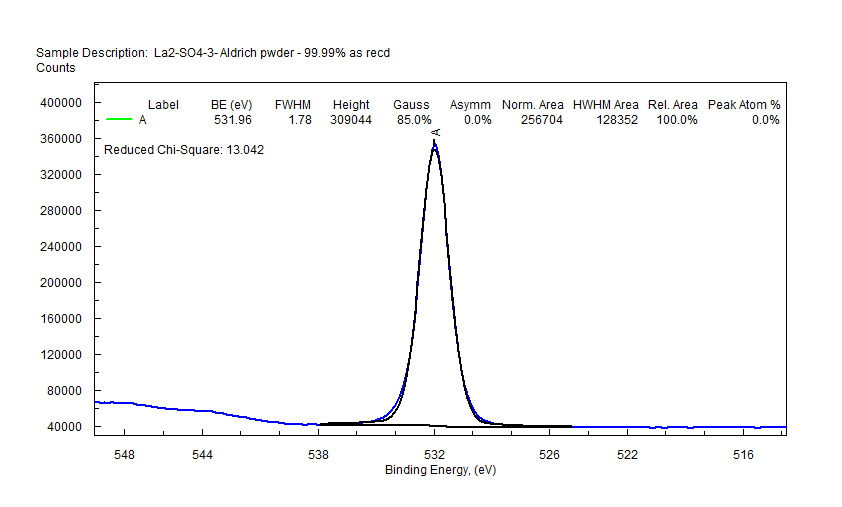 |
||
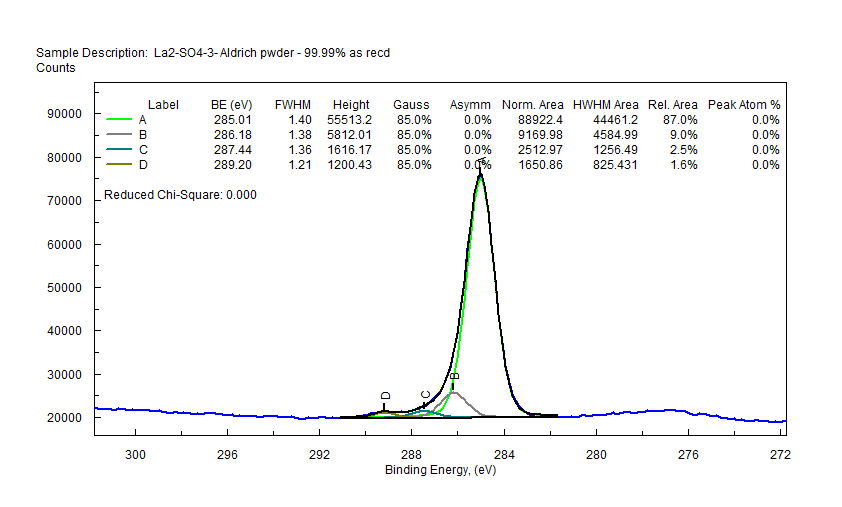 |

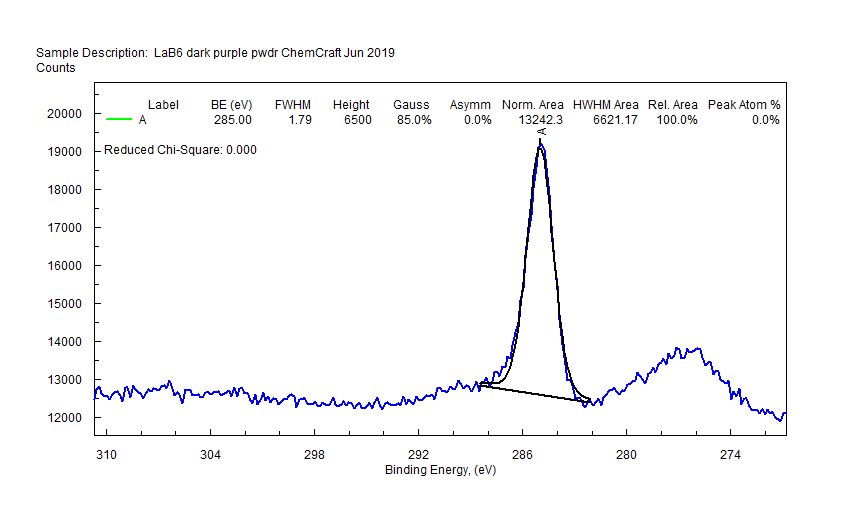
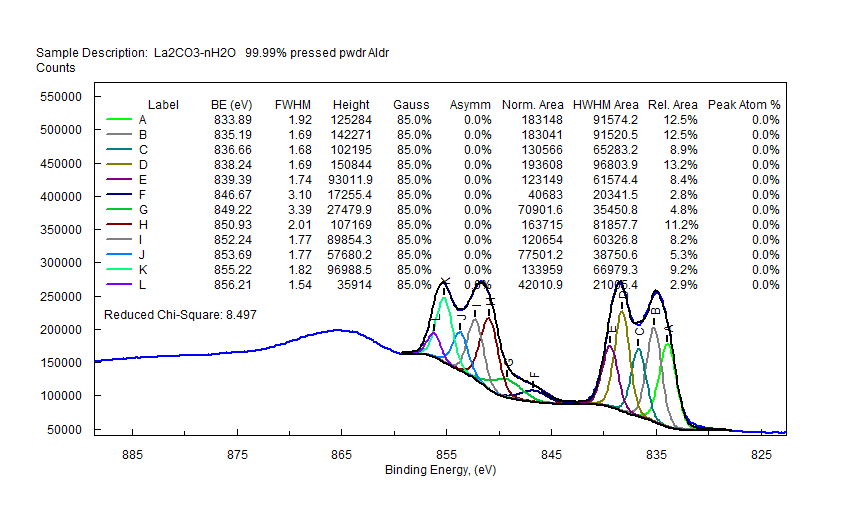
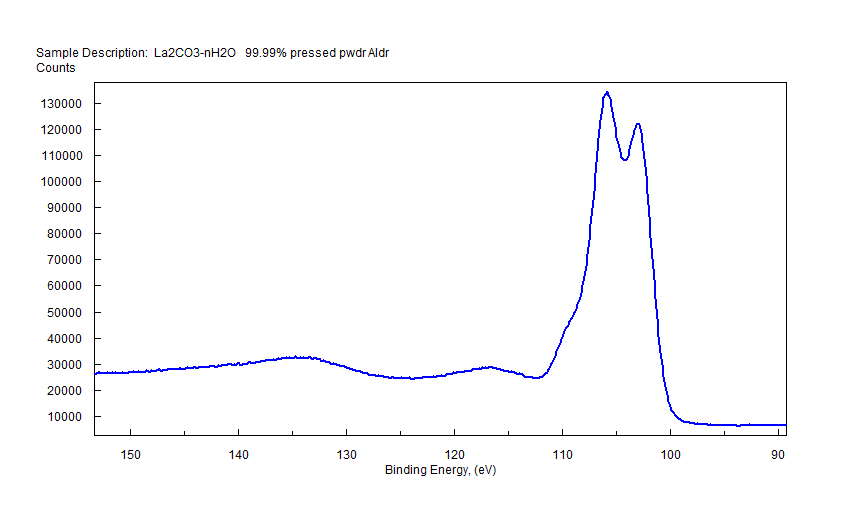
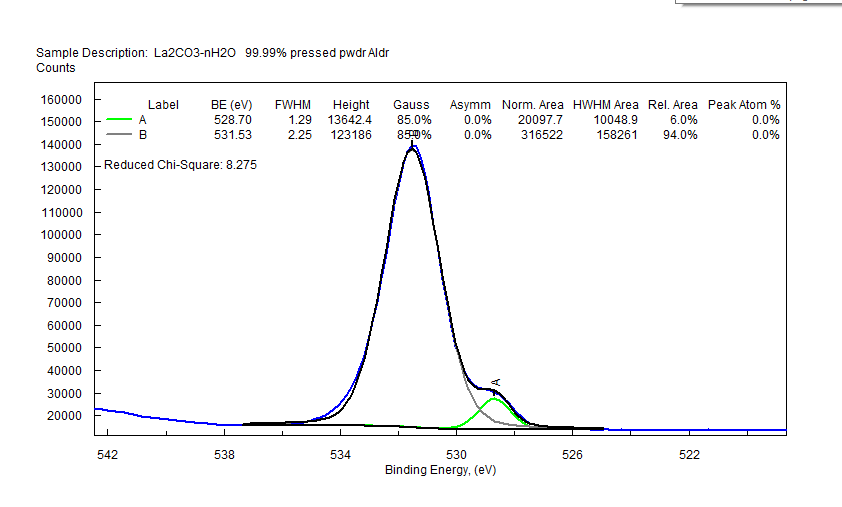
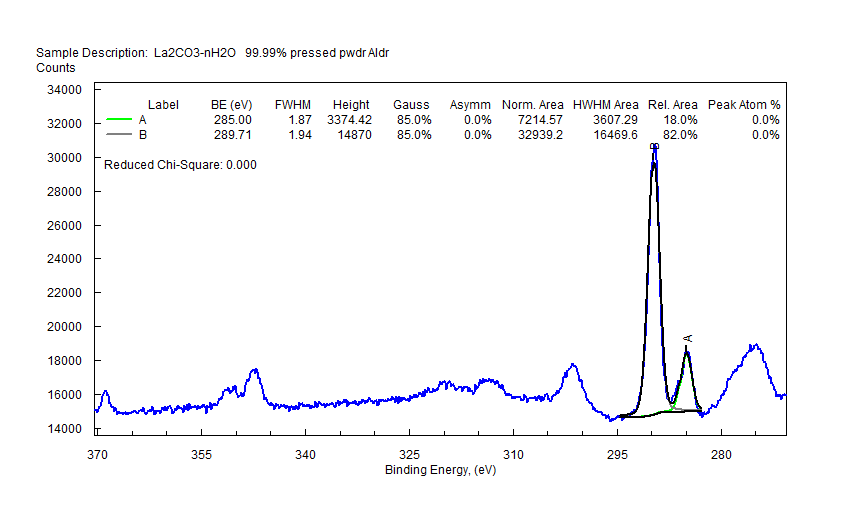
| Lanthanum (La) |  |
|
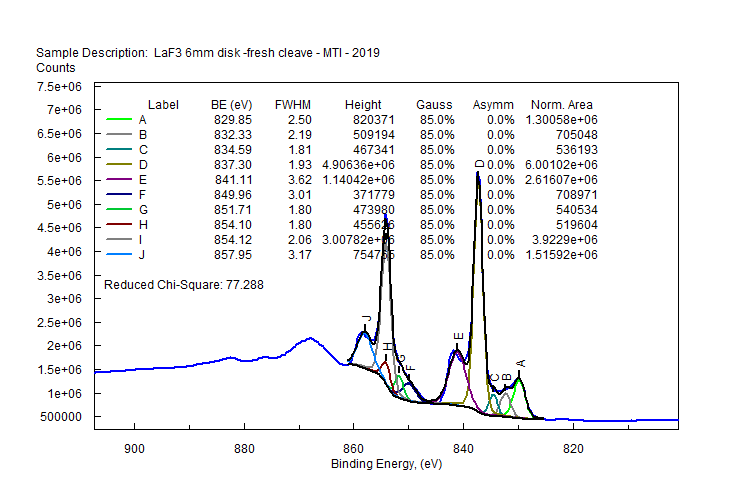 |
||
 |
||
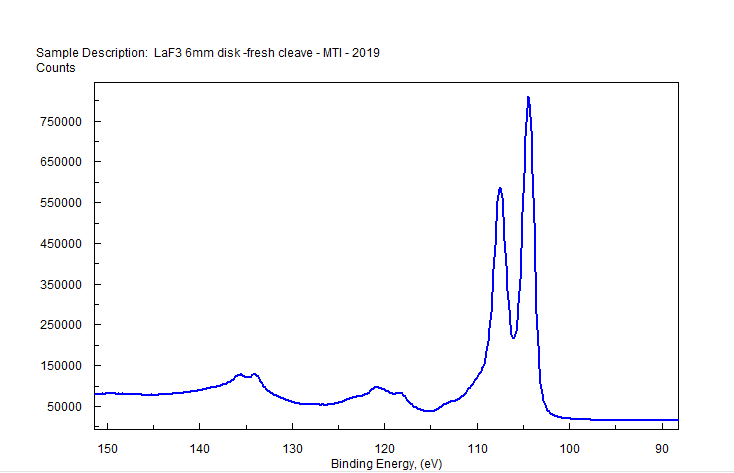 |
||
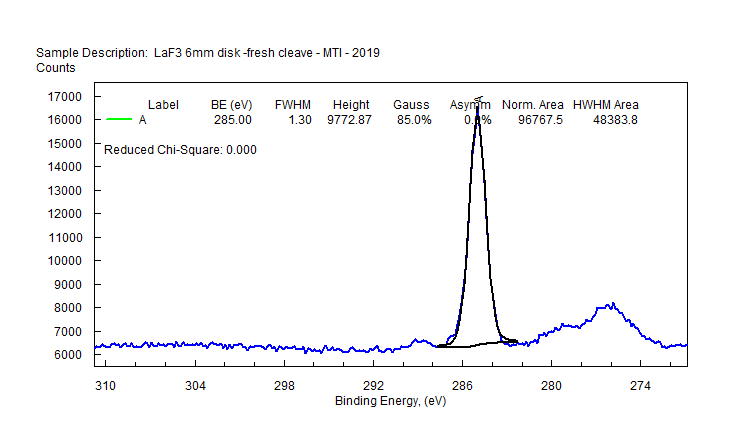 |
||
 |
||



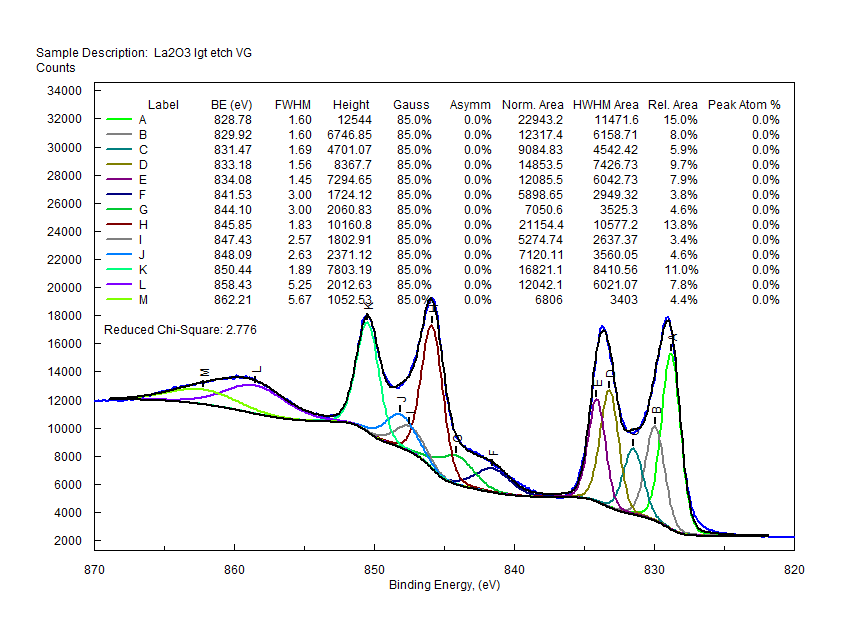
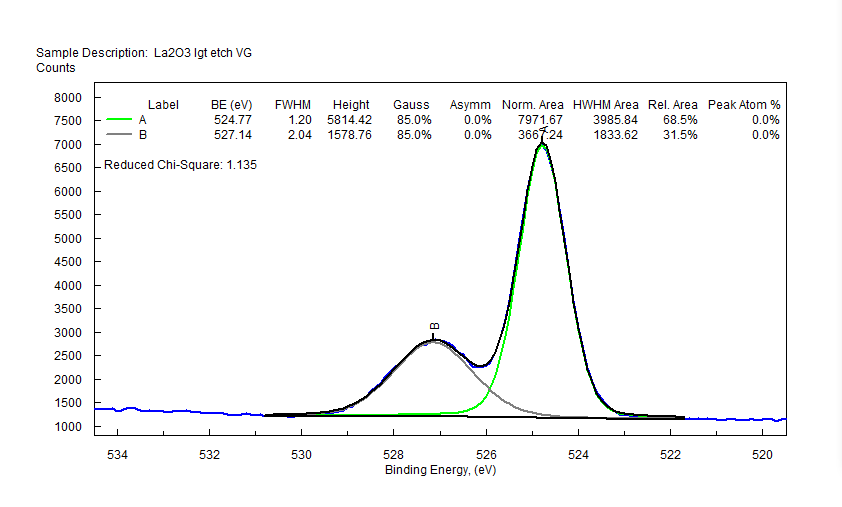
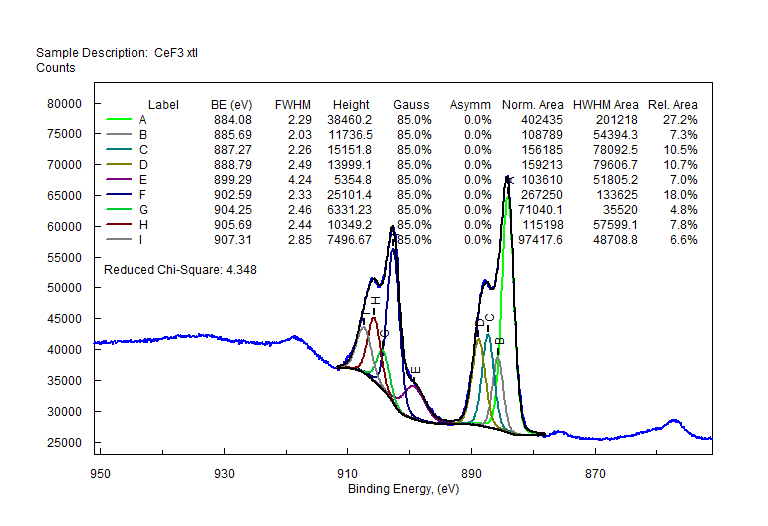
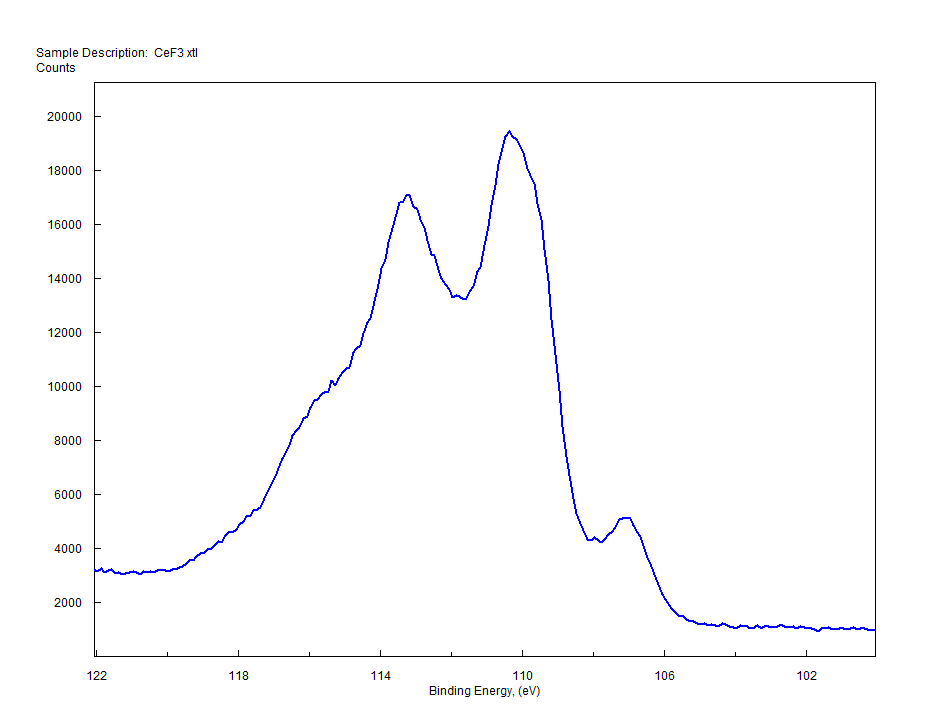
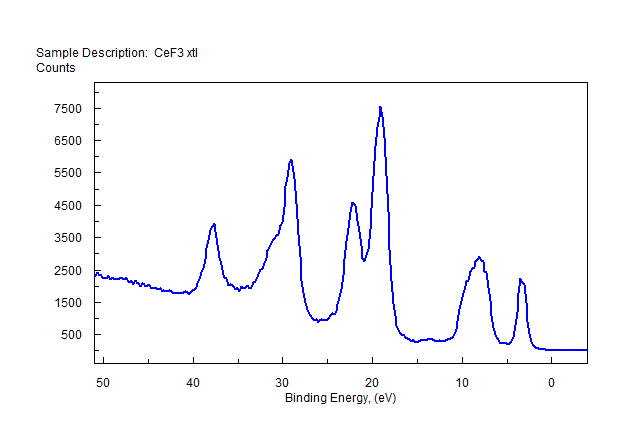
| Cerium (Ce) | 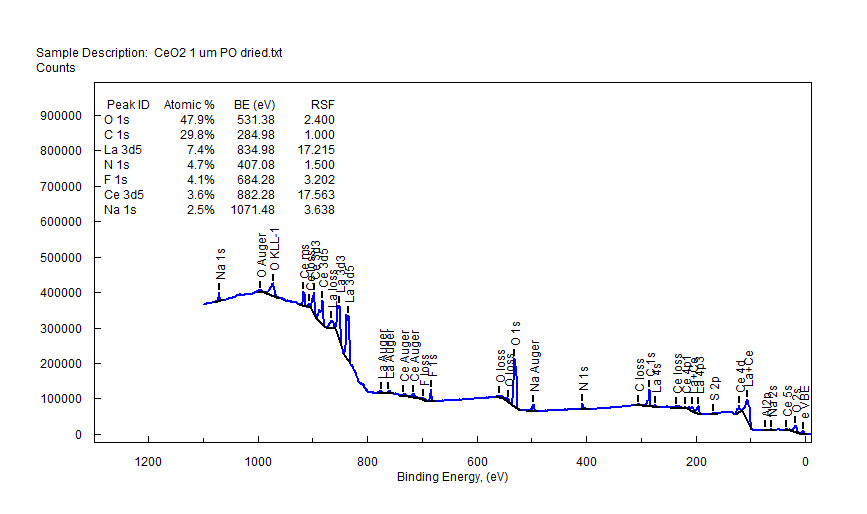 |
|
 |
||
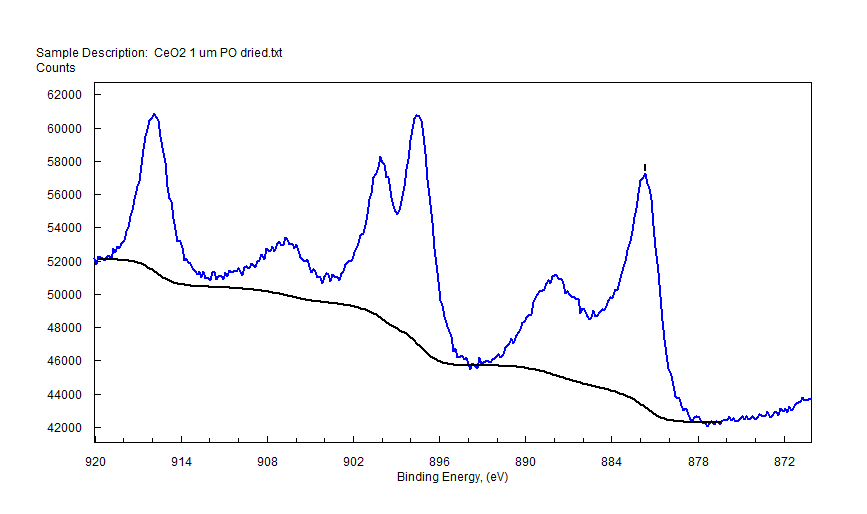 |
||
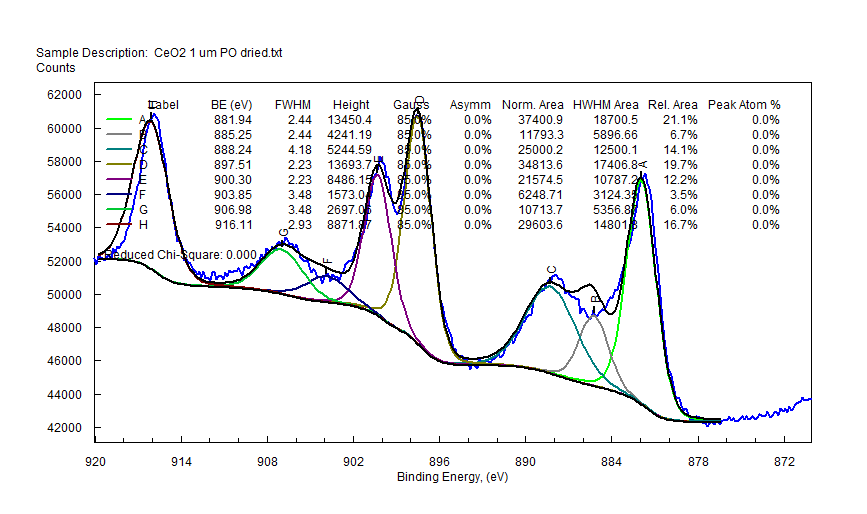 |

 |
||
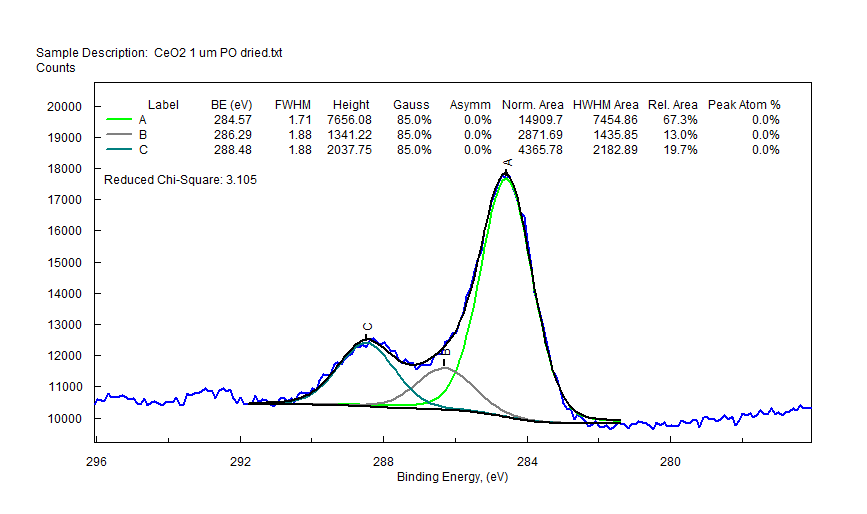 |
||
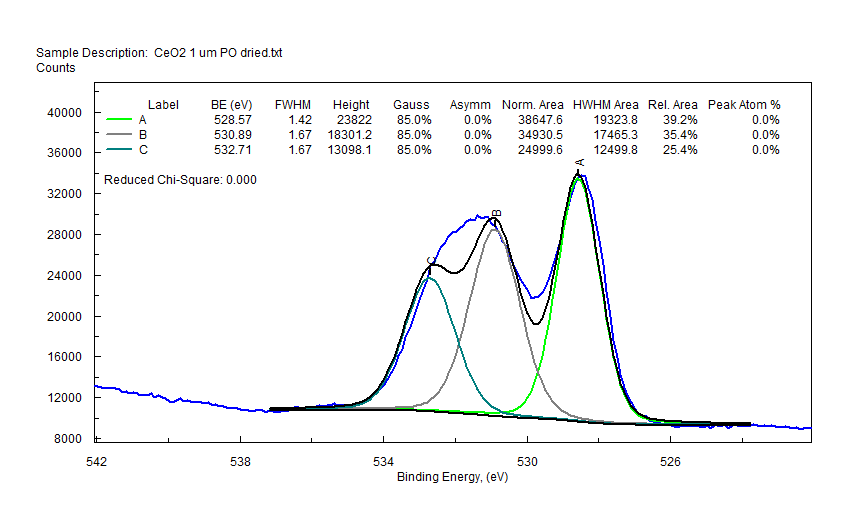 |
||
 |
||
 |
||
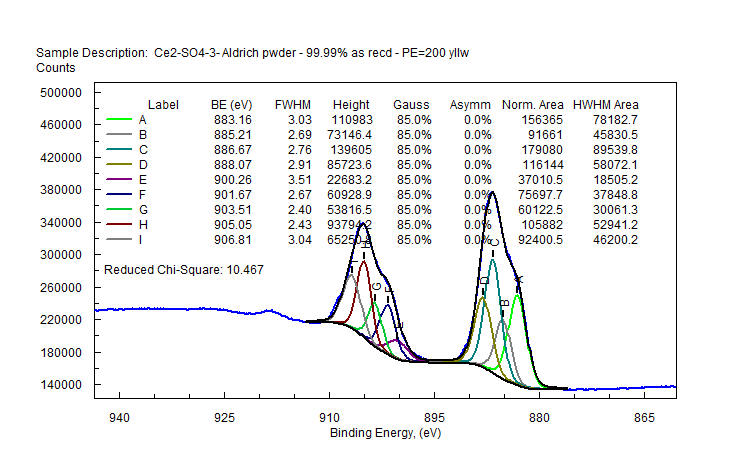 |

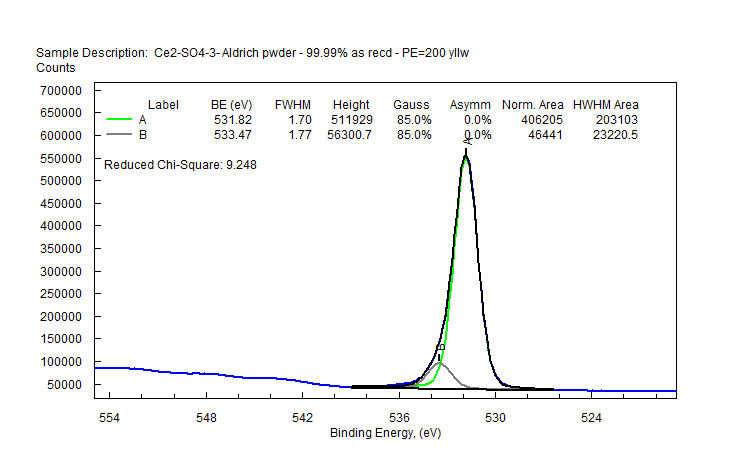 |
||
 |
||
 |
||
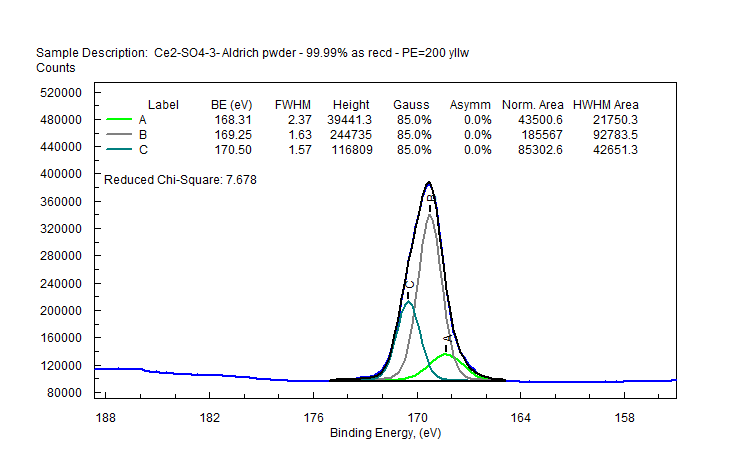 |
||
 |
||
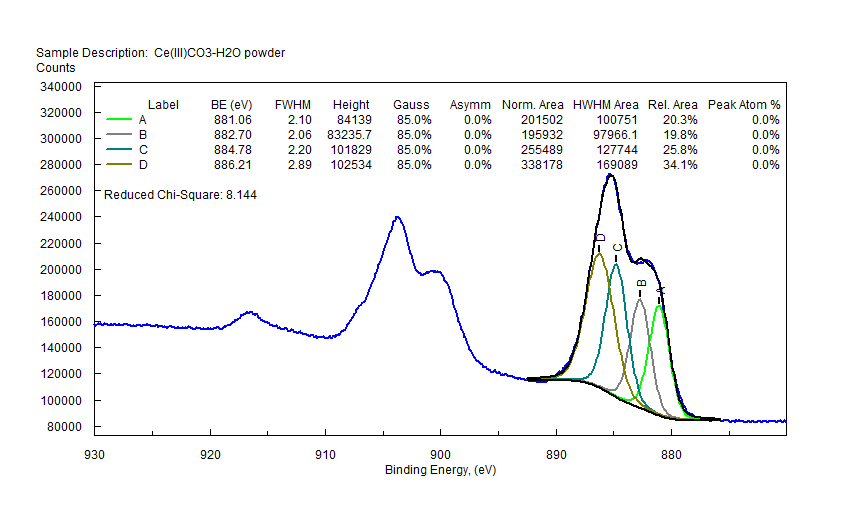 |
||
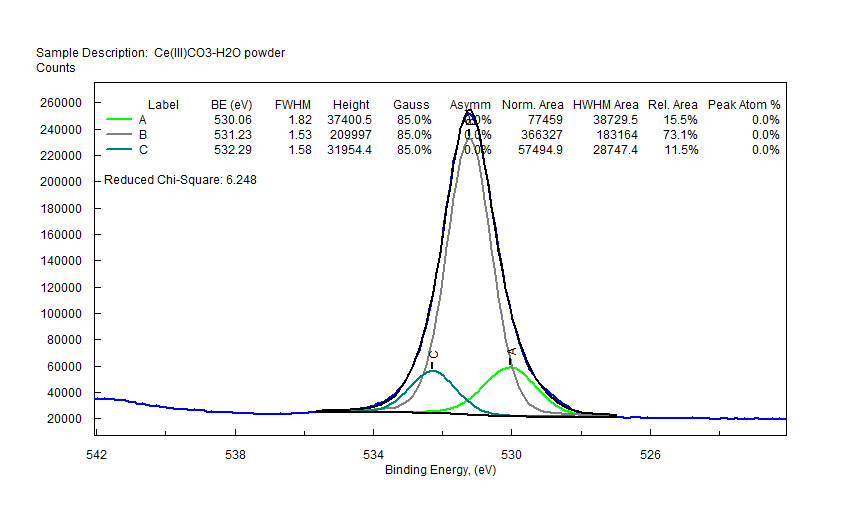 |
||
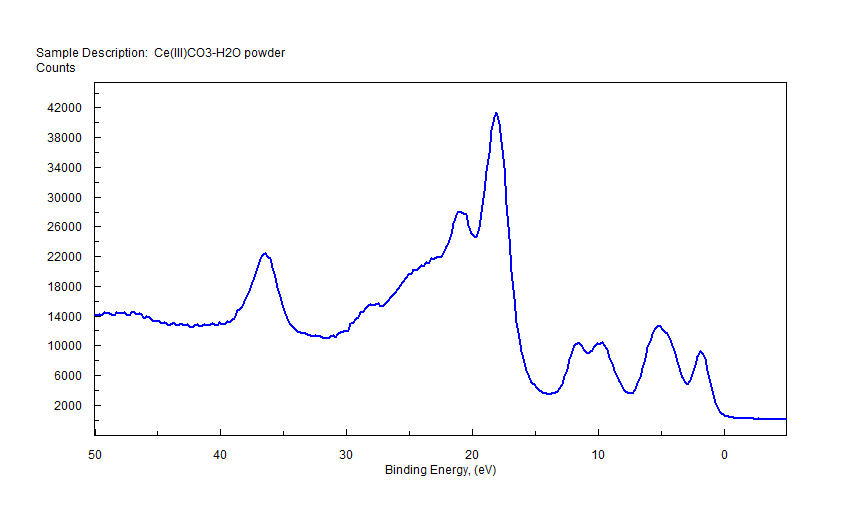 |
||
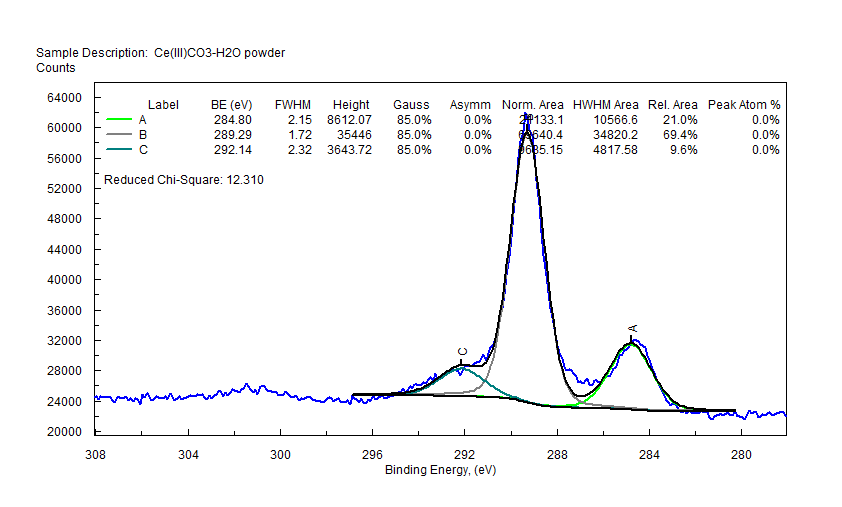 |
||
 |
 |
||
 |
||
 |
||
 |
||
 |
||
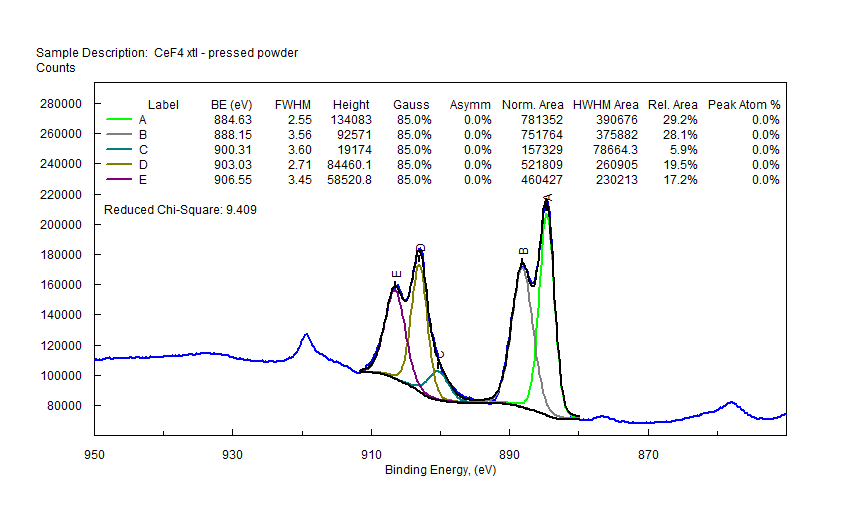 |
||
 |
||
 |
||
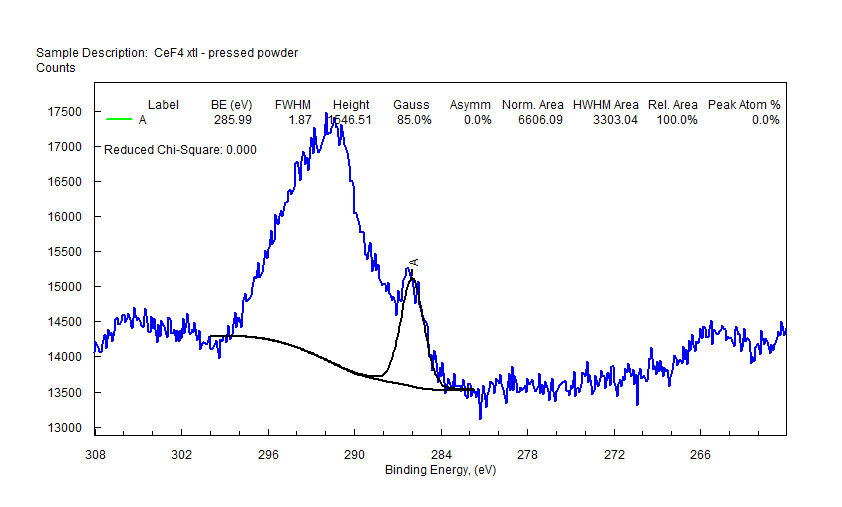 |



Multiplet Splittings
calculated by P. Bagus
Multiplet Splittings calculated by P. Bagus
Including XPS spectra for direct comparison
TiO2 and Ti2O3 Calculations
XPS Spectra
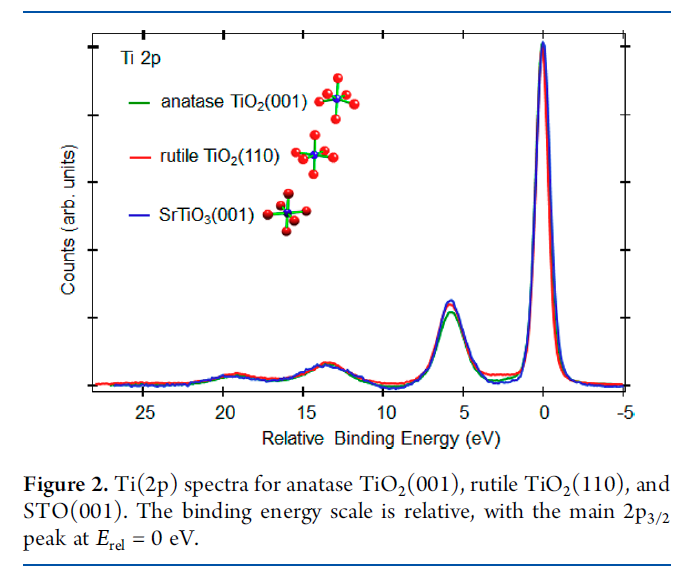

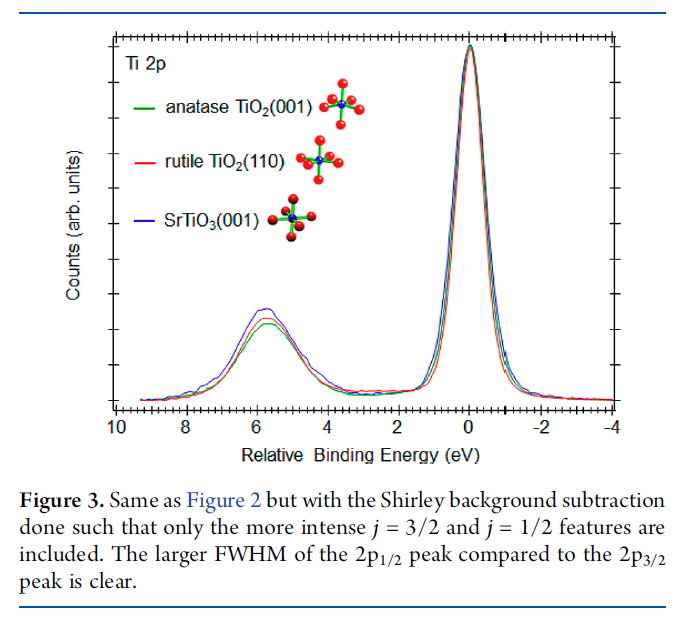







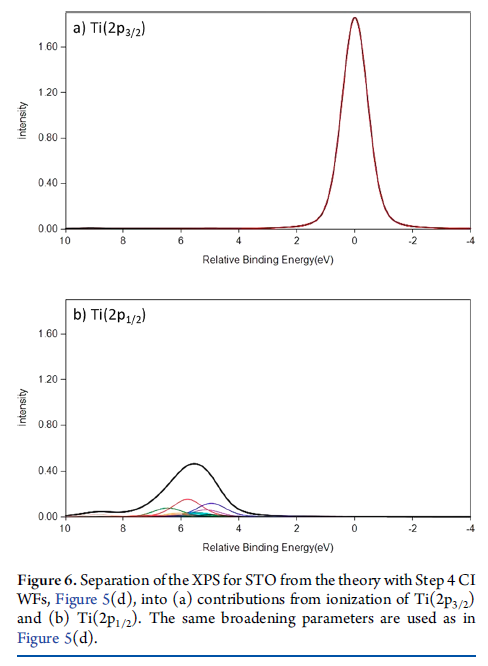

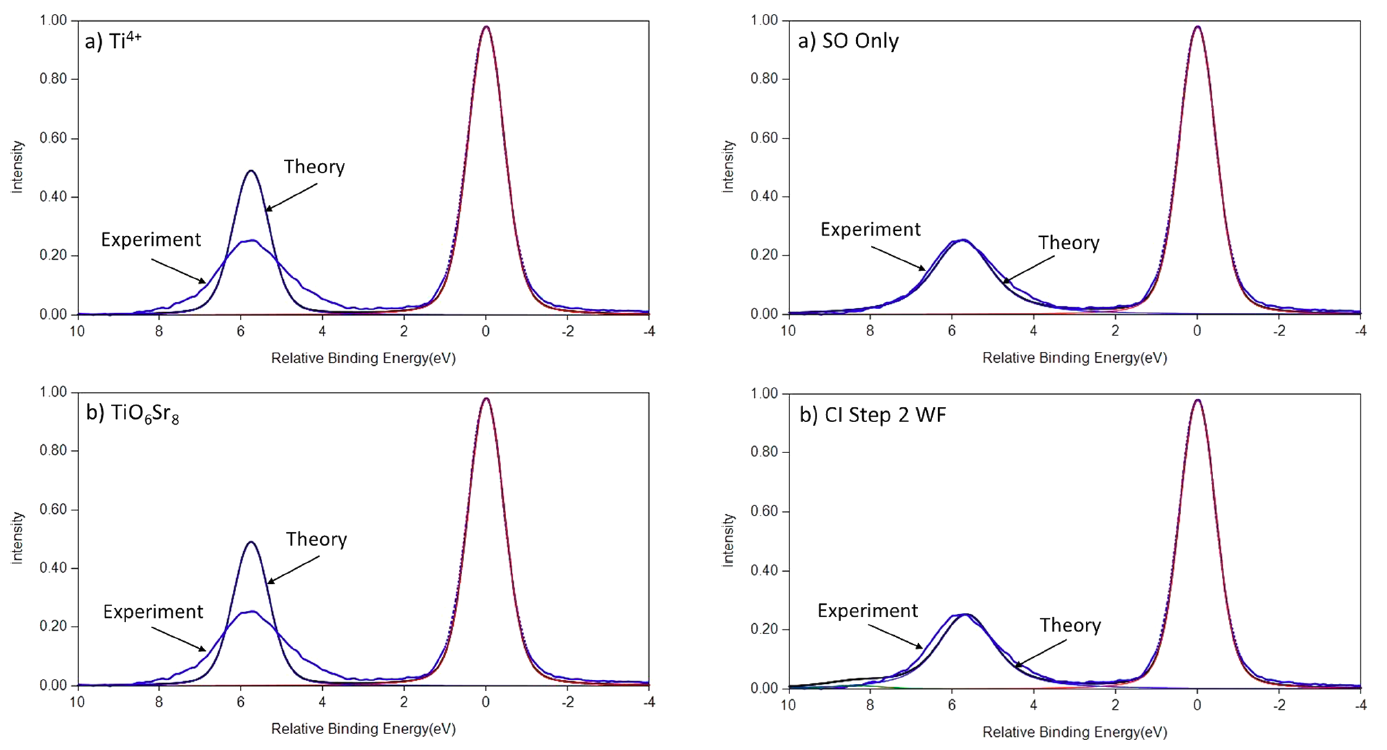

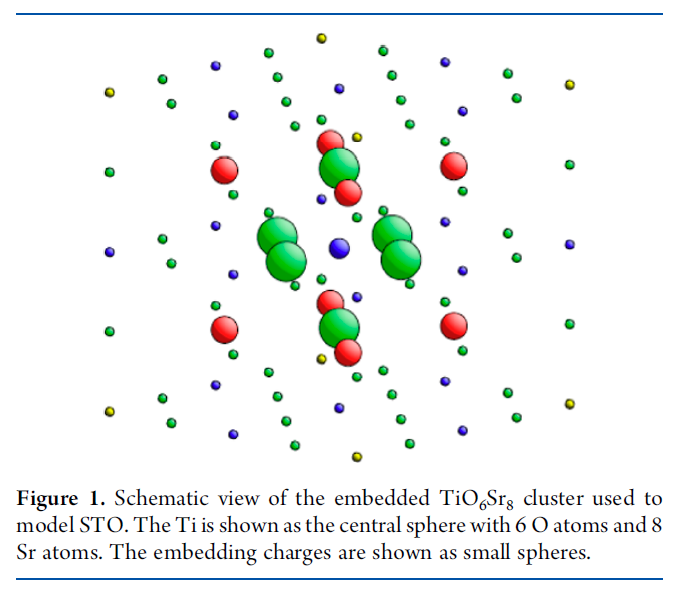




Cr2O3 Calculations
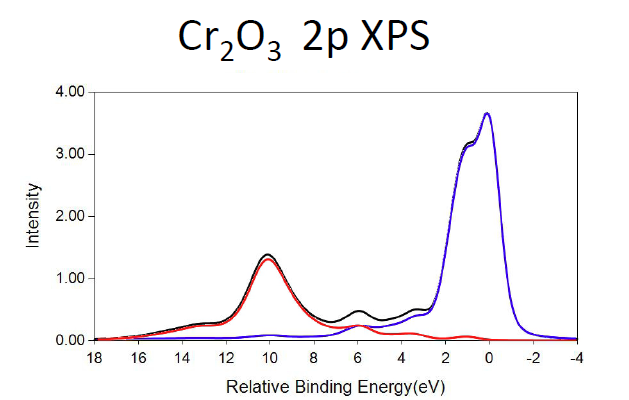

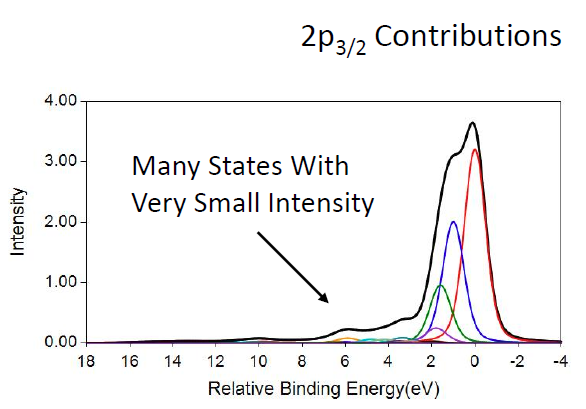



MnO Calculations


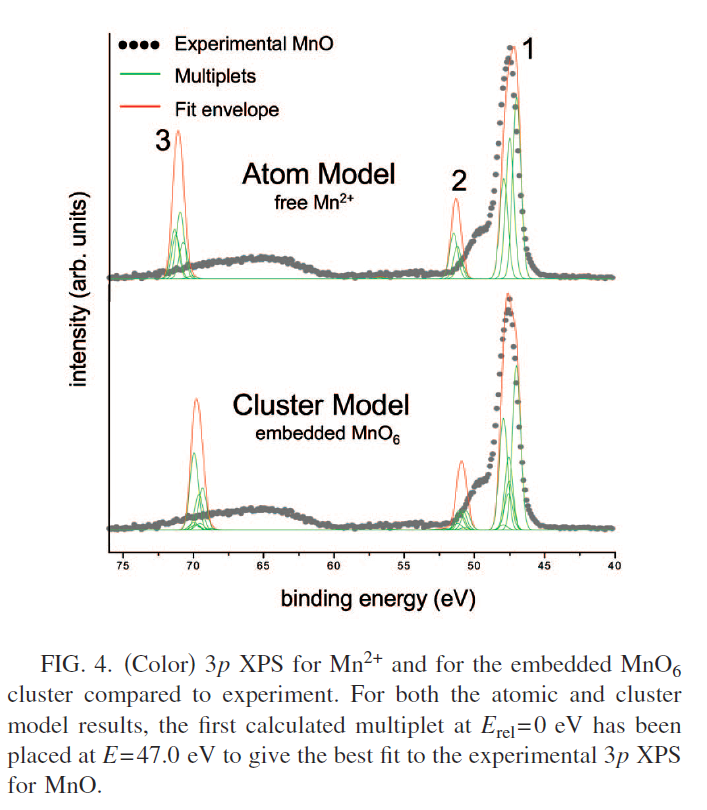

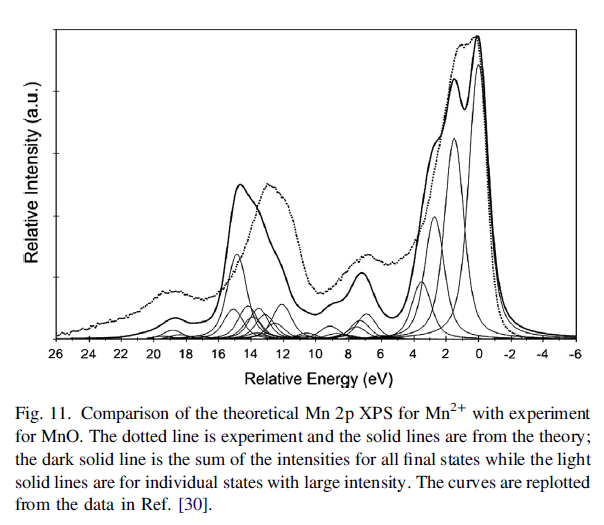


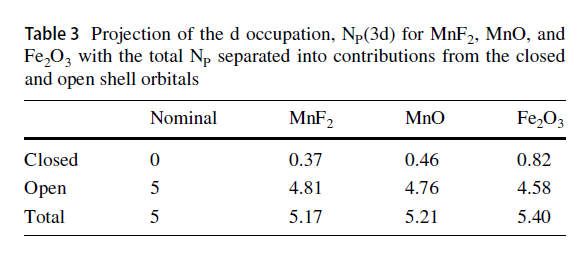

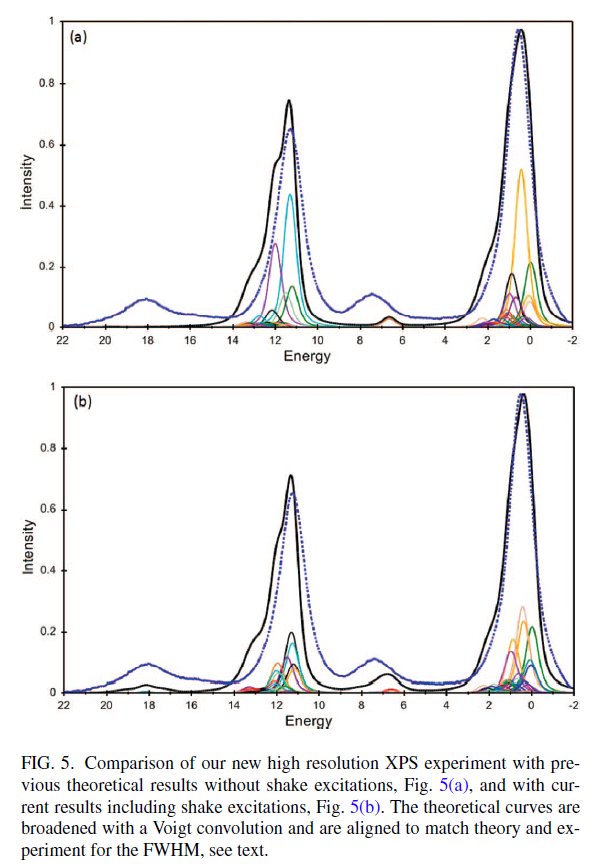
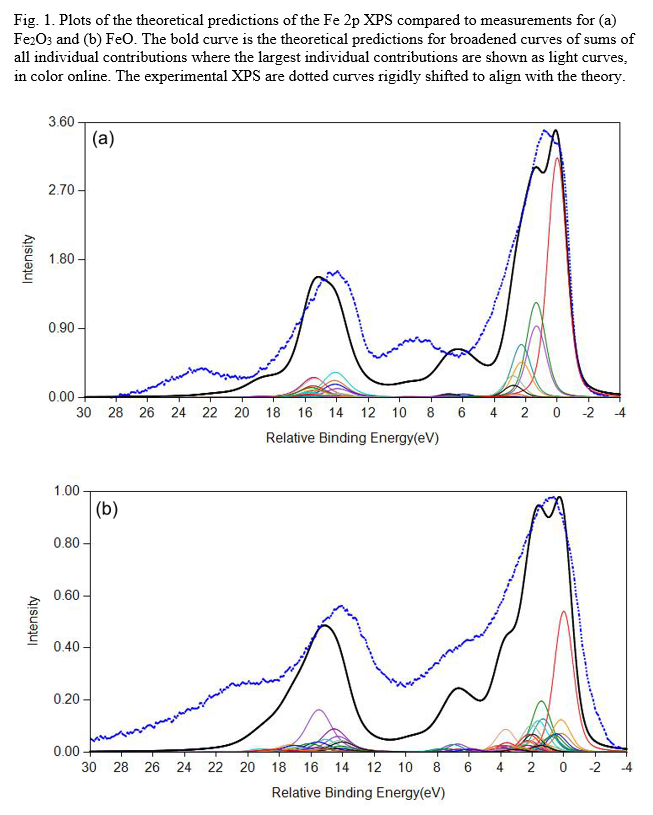
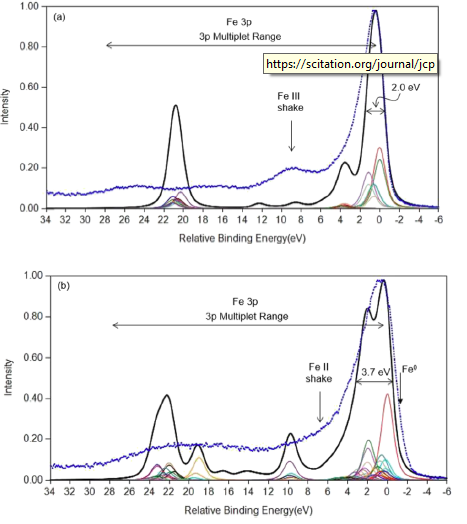
FeO and Fe2O3 Calculations


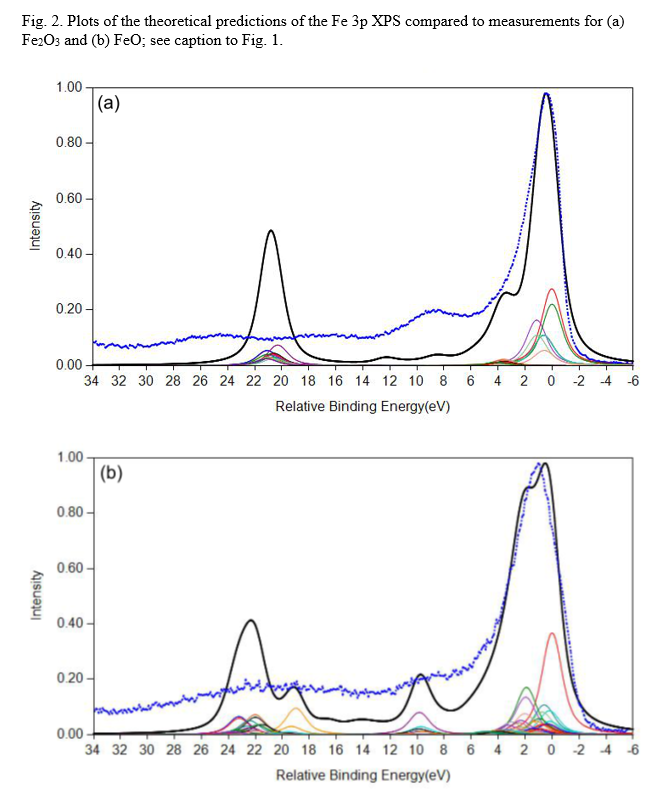

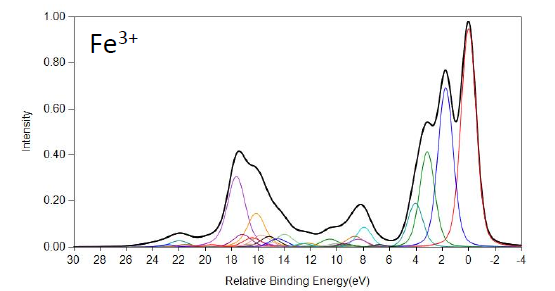

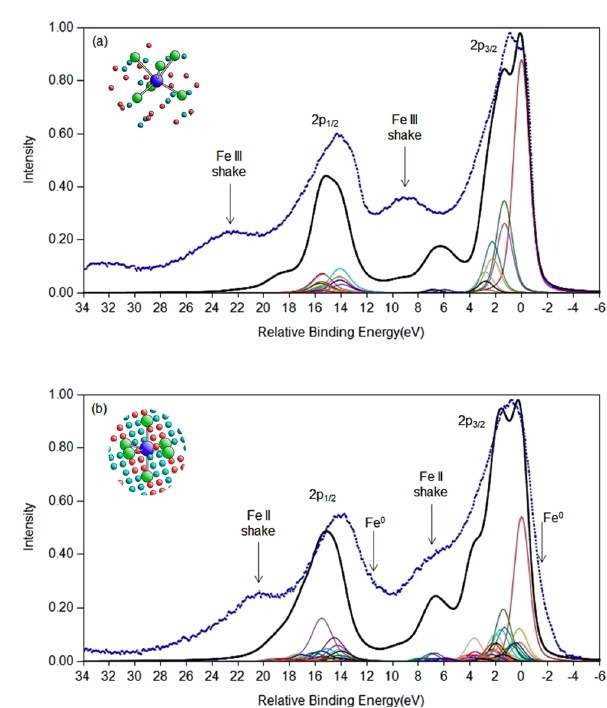

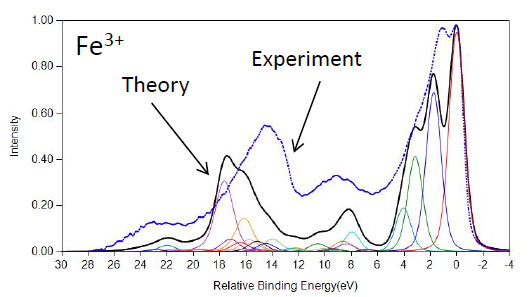

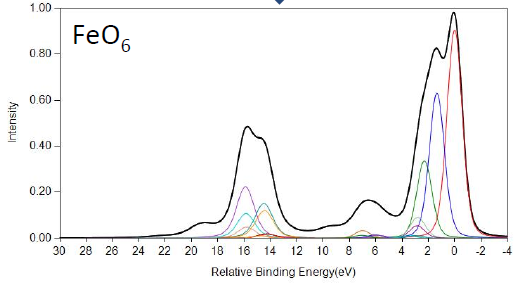

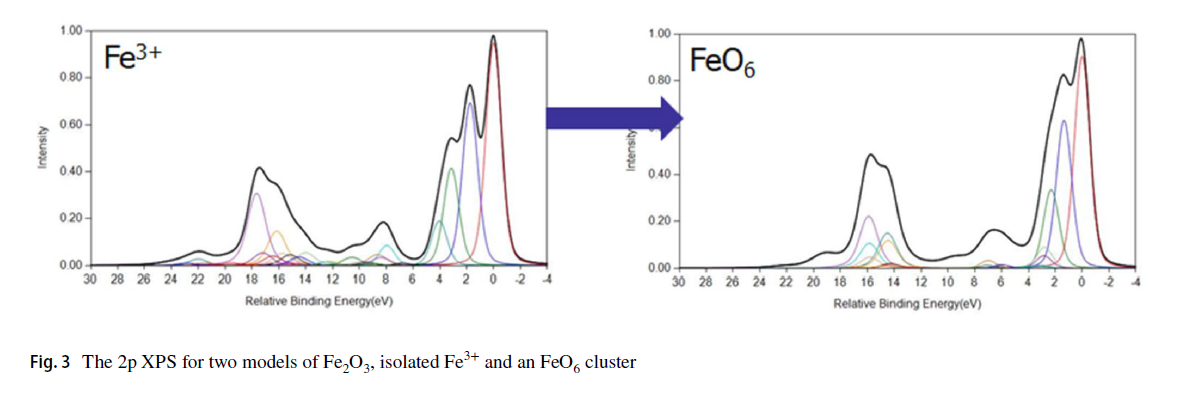

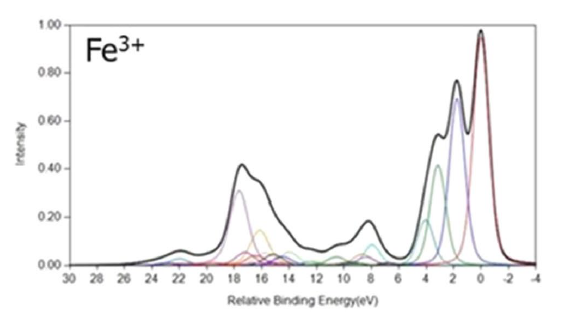

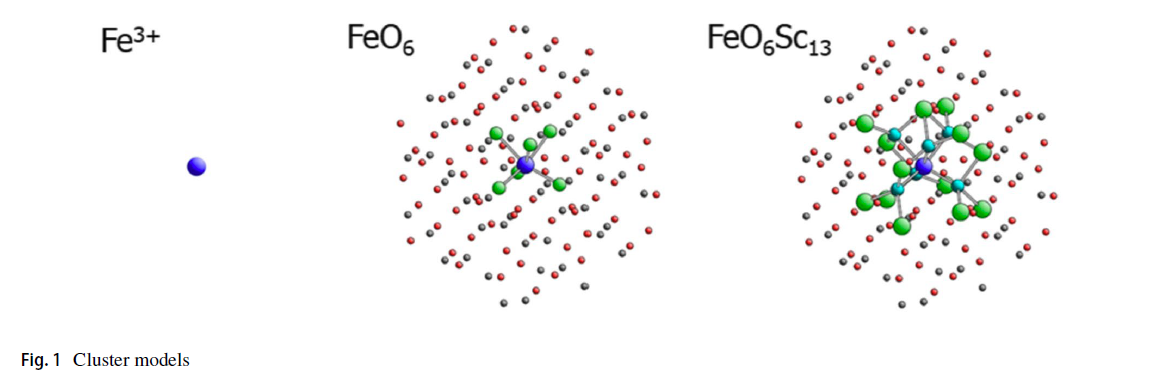


Cobalt Calculations
NiO Calculations
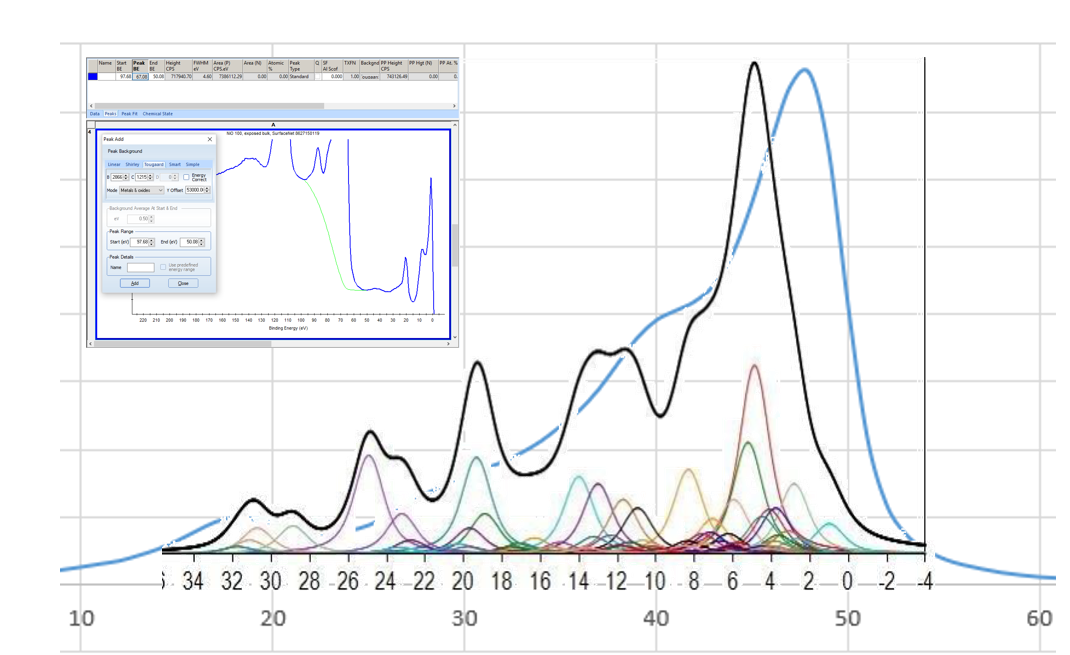

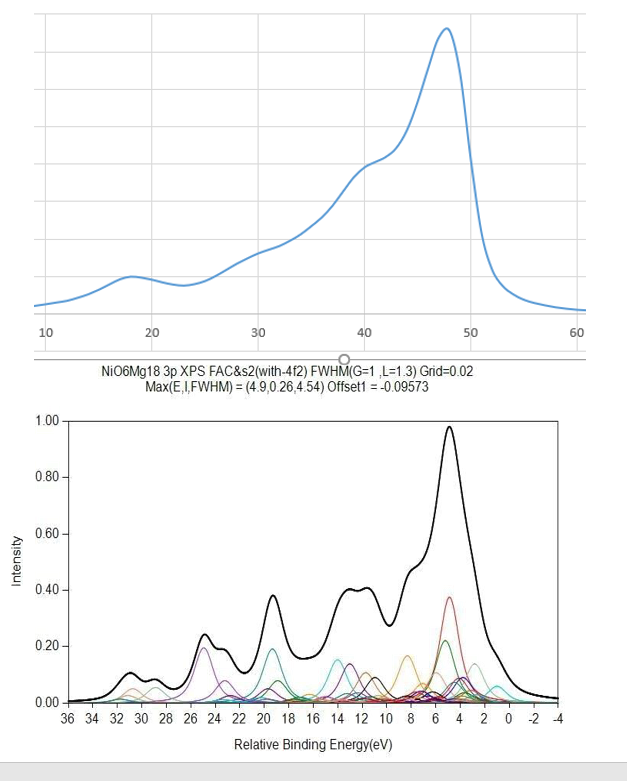

Copper Calculations
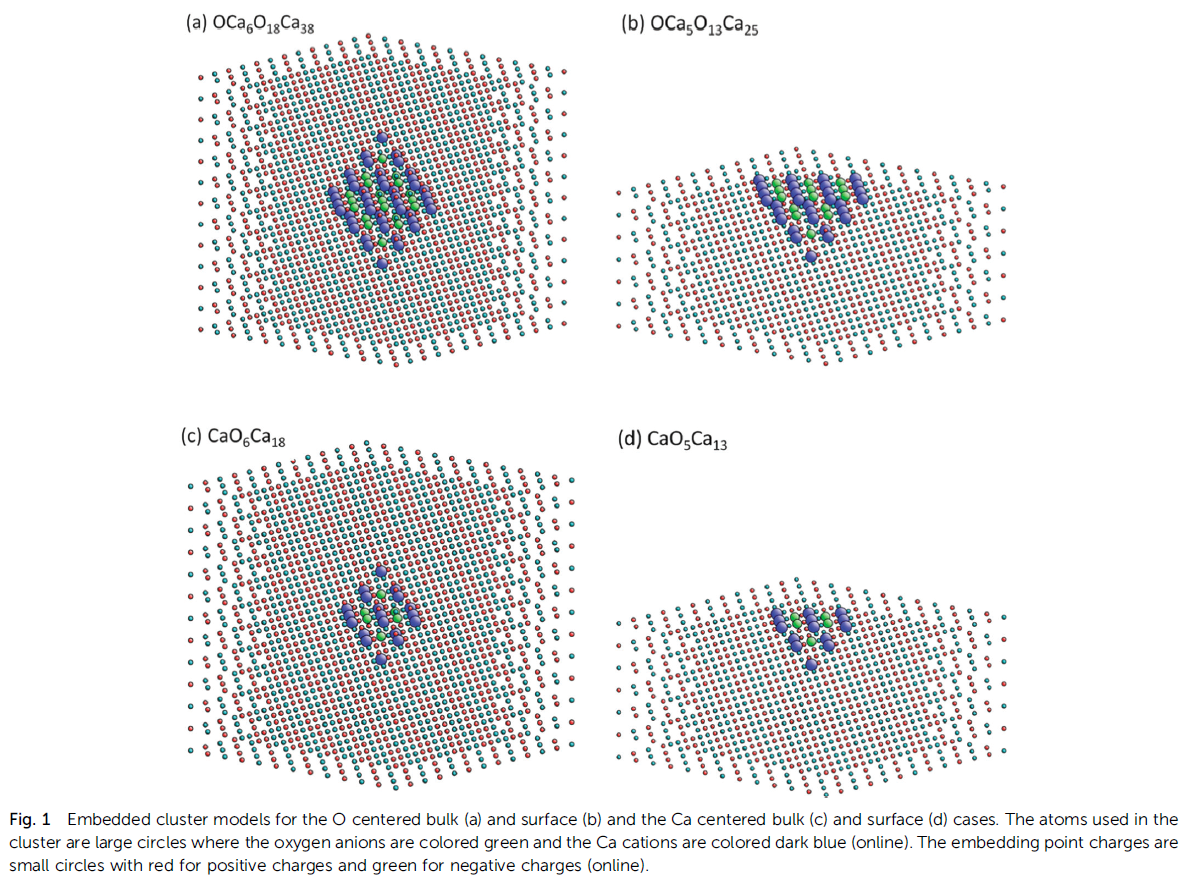
CaO Calculation
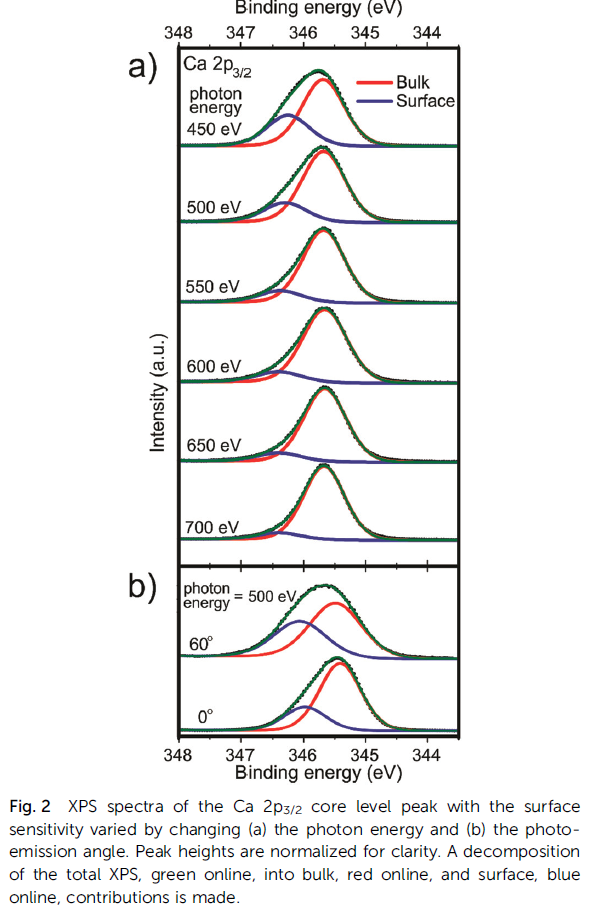

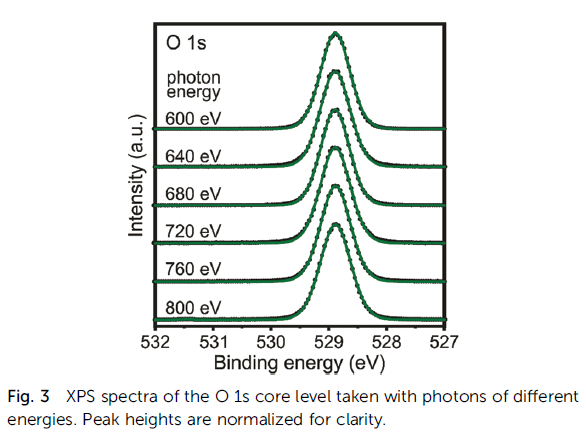


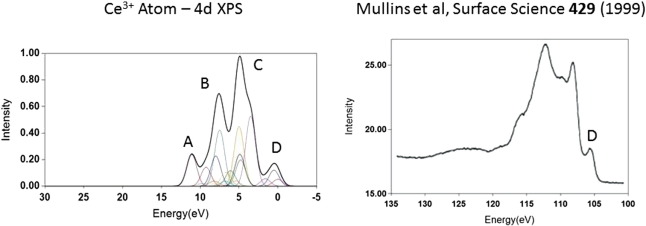
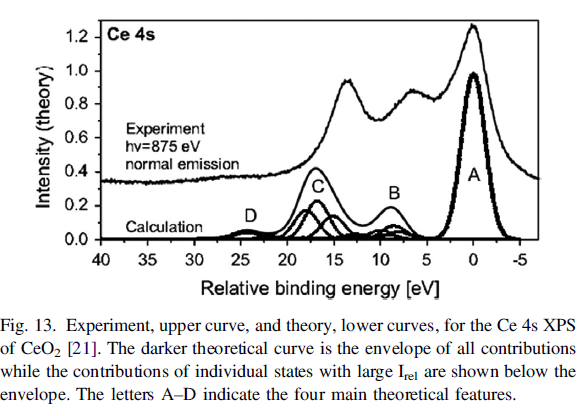
Cerium Oxides


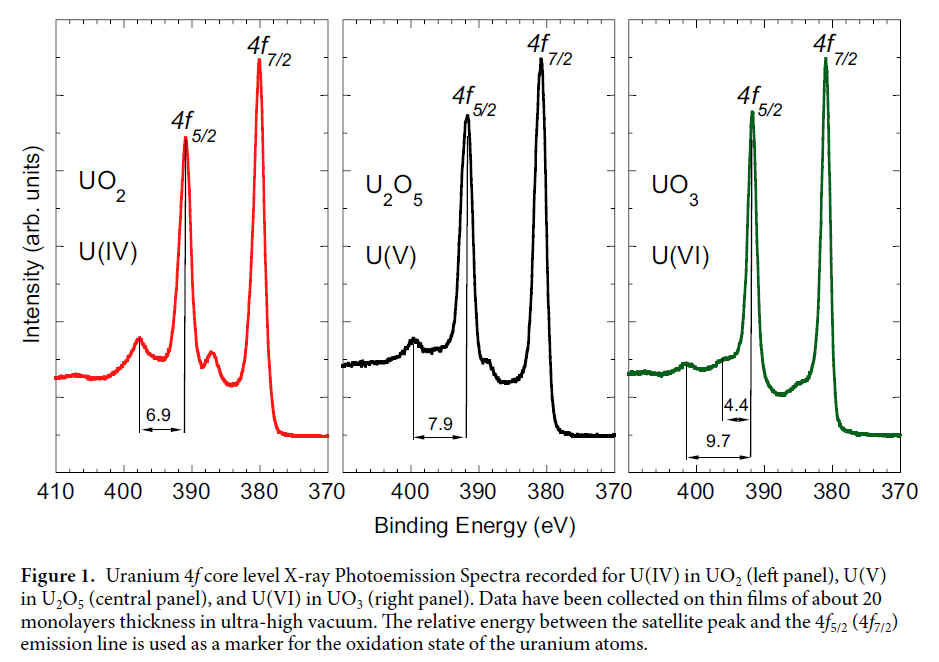
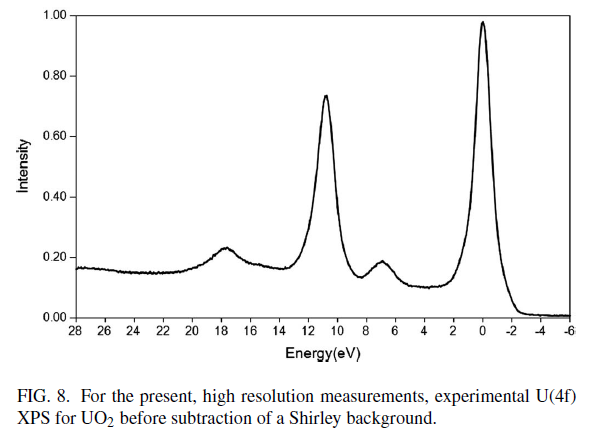
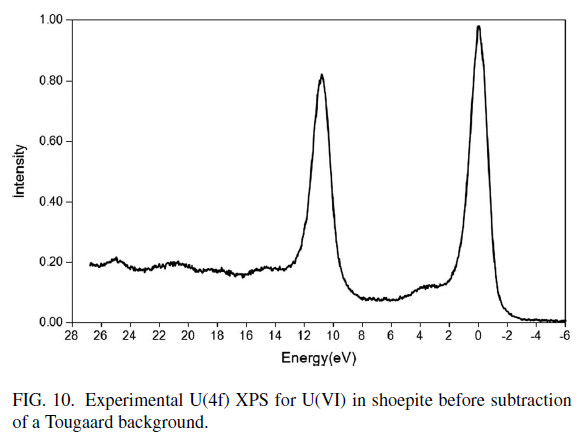
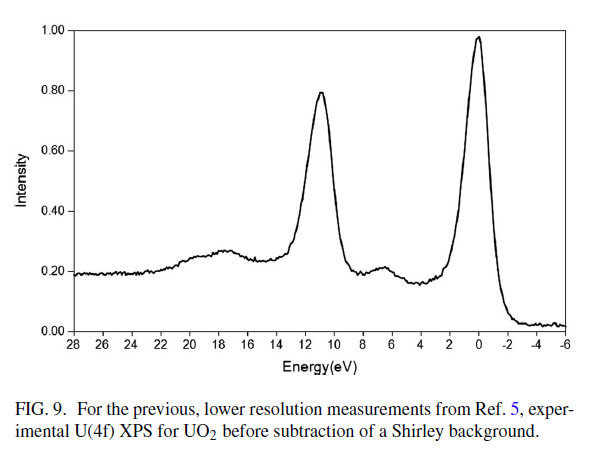
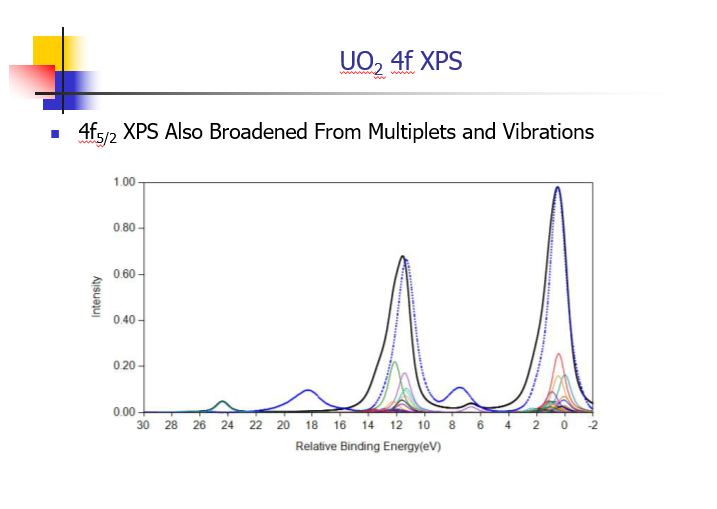
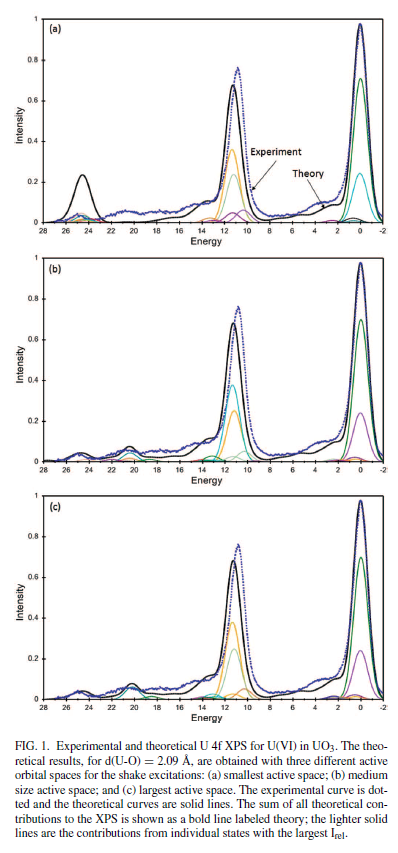
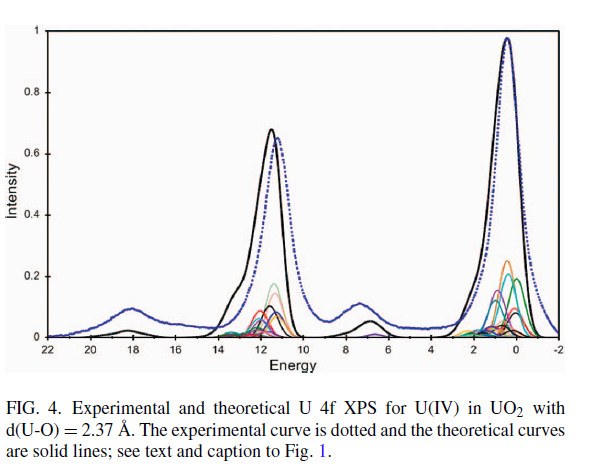
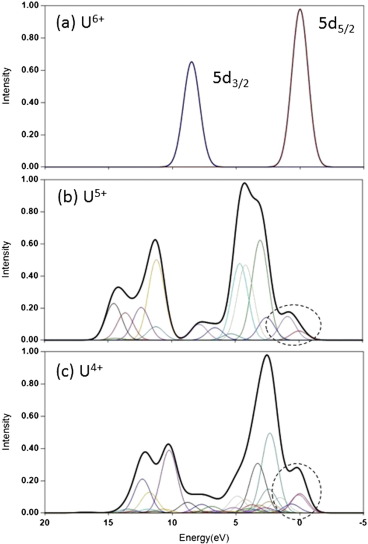
Uranium Oxides
XPS Spectra








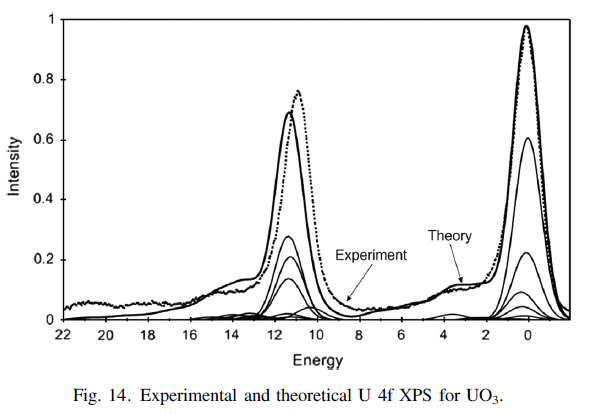


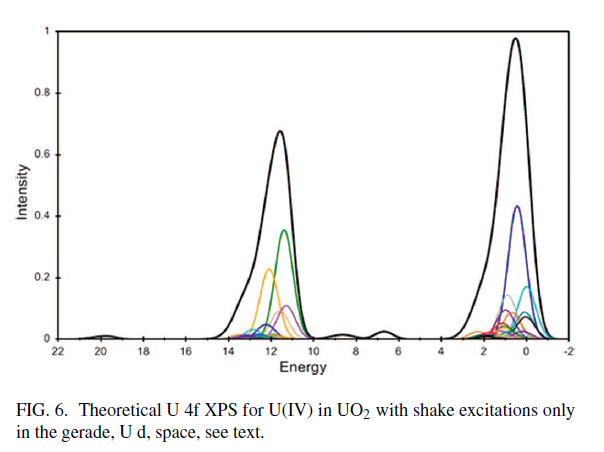

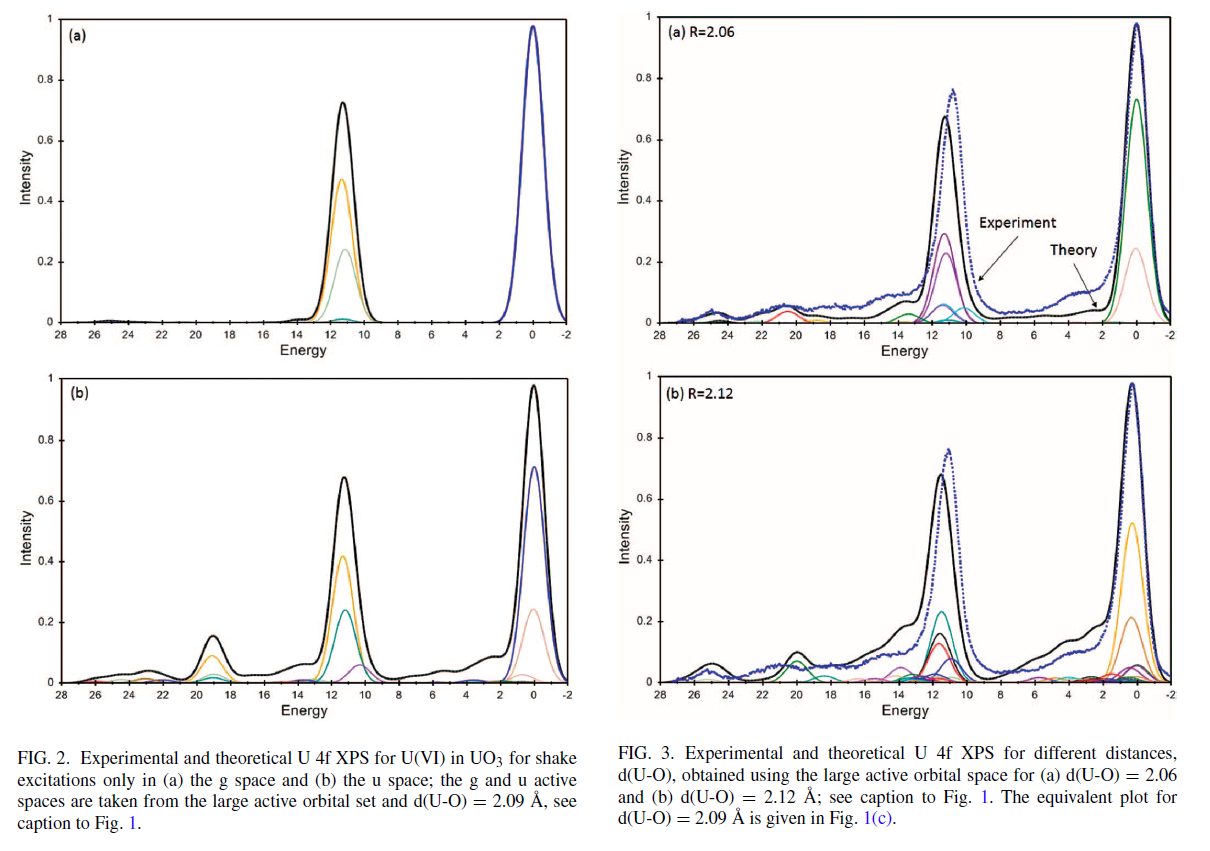

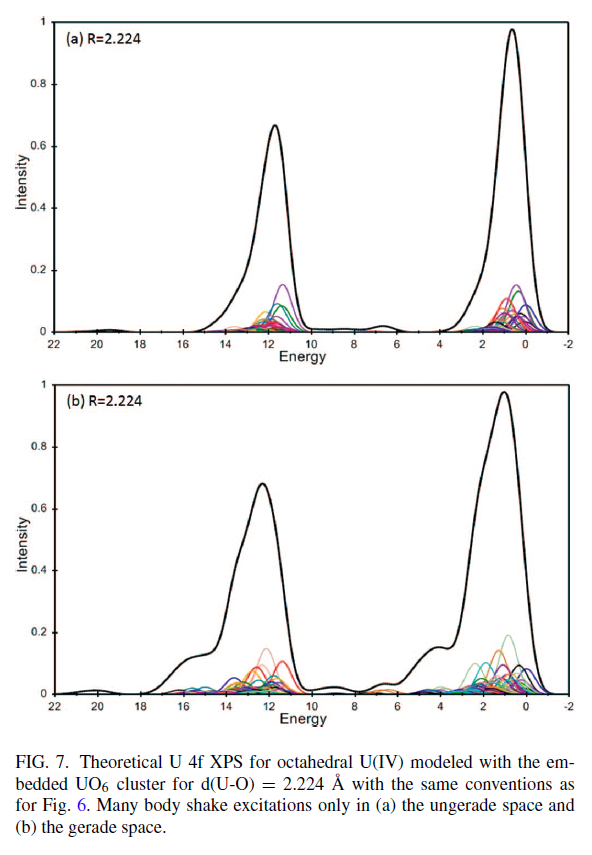

![]()

![]()